
The PD-1/PD-L1 Axis in the Biology of MASLD

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Innate and Adaptative Immunity in the MASLD
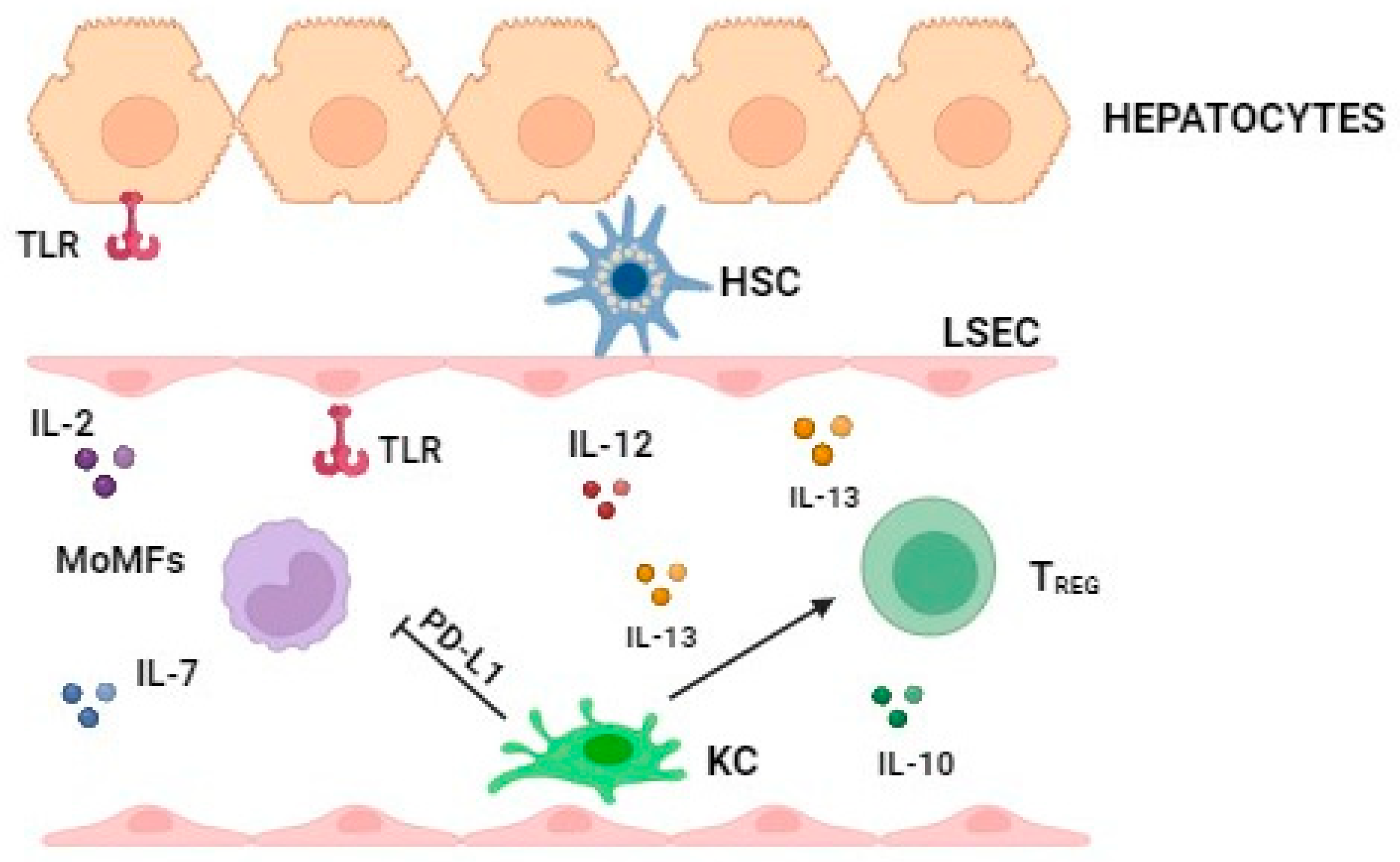
2.1. The Major Immune Cell Types in the Phisiological Status and in the Early Stage of MASLD
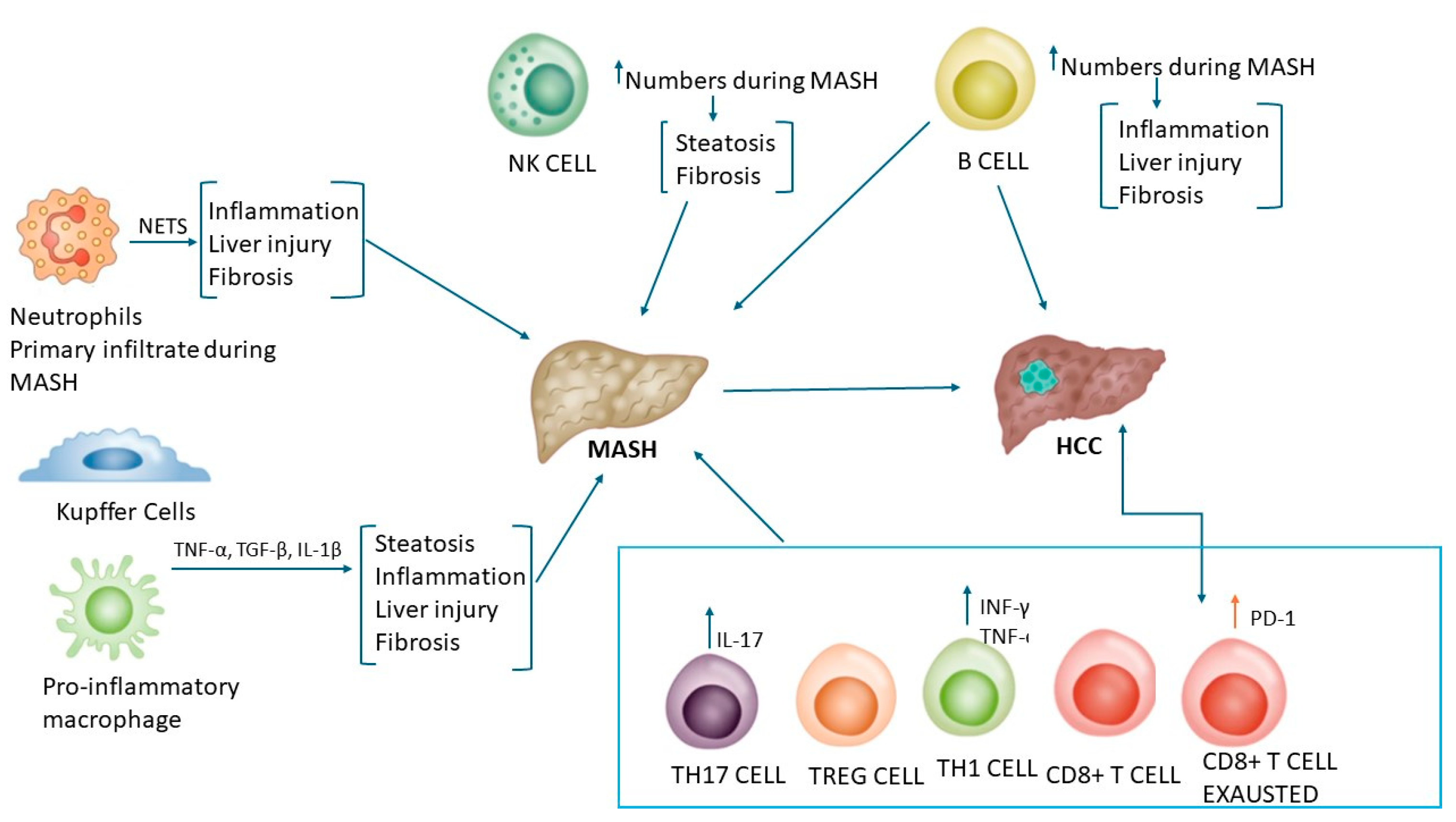
2.2. Role of the Major Immune Cell Types in the Late Stage of MASLD
2.3. Role of the Adaptative Immune Response in MASH Progression
2.3.1. The Humoral Immunity: B Lymphocytes
2.3.2. The Cellular Immunity: T Lymphocytes
3. Impact of PD-1/PD-L1 in Chronic Metabolic Diseases
3.1. The PD-1/PD-L1 Immune Pathway and the Role of CTLA-4/B7 and TIGIT/CD112 Co-Inhibitor Receptors
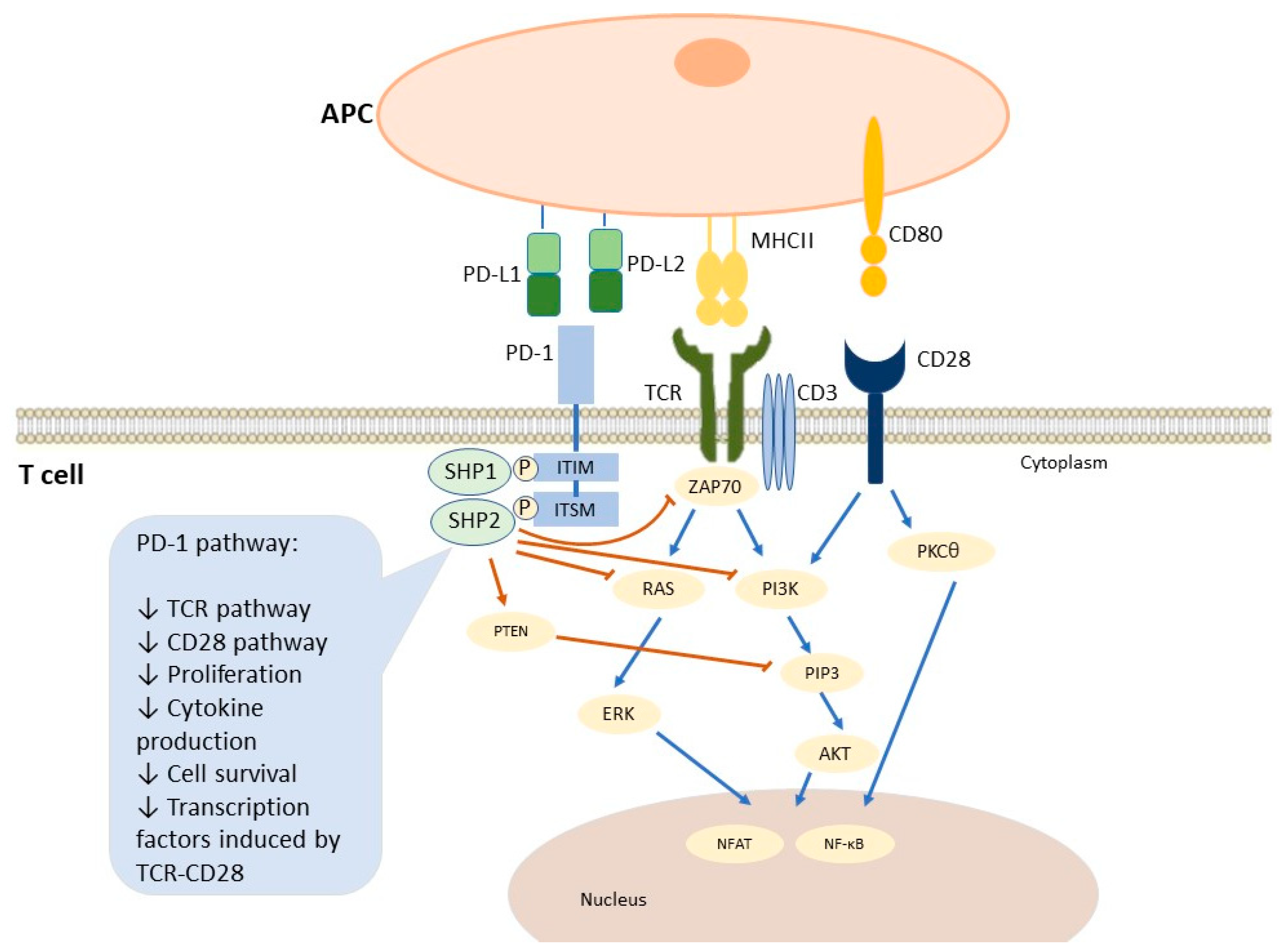
3.1.1. PD-1/PD-L1 Immune Pathway
3.1.2. CTLA-4/B7: A Co-Inhibitor Receptor of Immune Response
3.1.3. TIGIT/CD112: A Co-Inhibitor Receptor of Immune Response
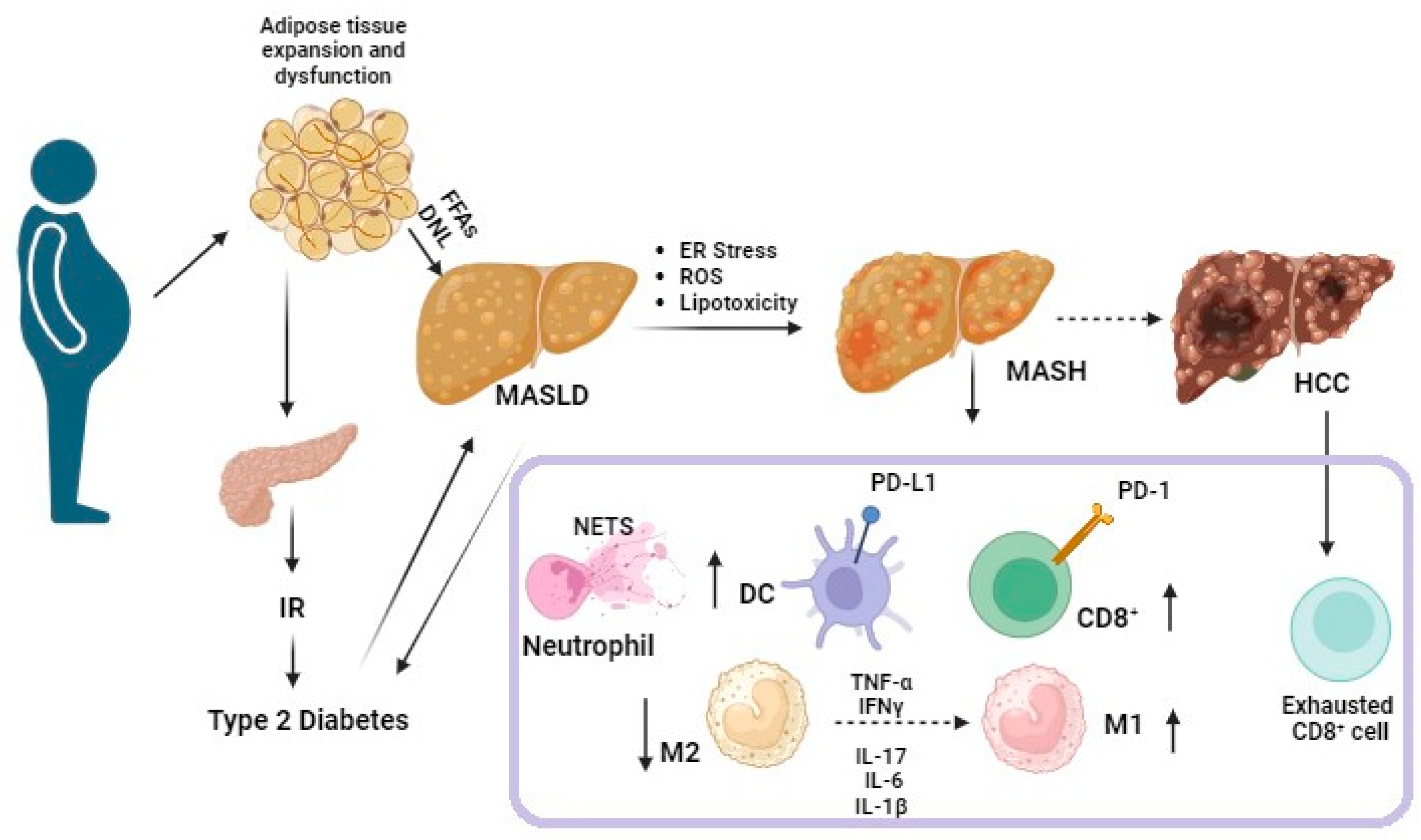
3.2. PD-1/PD-L1 Axis in Obesity
3.3. PD-1/PD-L1 Axis in Type 2 Diabetes
3.4. The PD-1/PD-L1 Axis in MASLD
Pro- and Anti-Inflammatory Effects of PD-1/PD-L1 Axis in MASLD Progression
4. Potential Role of PD-1/PD-L1 as Biomarkers in MASLD
5. The Rationale of Immunotherapy in MASLD
6. Limitations
7. Conclusions
8. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cell |
| CCR | Chemokine (C-C motif) ligand (CCL) 2/C-C chemokine receptor |
| CD | Cluster of differentiation |
| CTLA-4 | Cytotoxic T Lymphocyte Antigen 4 |
| DC | Dendritic cell |
| HCC | Hepatocellular carcinoma |
| HSC | Hepatic stellate cells |
| ICI | Immune Checkpoint Inhibitors |
| Ig | Immunoglobulin |
| ILC2 | Innate lymphoid type 2 cell |
| IFN | Interferon |
| IL | Interleukin |
| IR | Insulin resistance |
| KC | Kupffer cell |
| LSEC | Liver sinusoidal endothelial cell |
| MoMf | Monocyte-derived macrophage |
| MASLD | Metabolic-associated fatty liver disease |
| MASH | Metabolic dysfunction-Associated Steatohepatitis |
| NASH | Nonalcoholic steatohepatitis |
| NETs | Neutrophil Extracellular Traps |
| NK | Natural killer |
| NKT | Natural killer T |
| OCA | Obeticholic acid |
| PPAR | Peroxisome proliferator-activated receptor |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| T2D | Type 2 diabetes mellitus |
| TGF | Transforming growth factor |
| TH | T helper |
| TIGIT | T-cell Ig and ITIM domain |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| T Reg | T regulatory |
| WAT | White adipose tissue |
References
- Vitale, A.; Svegliati-Baroni, G.; Ortolani, A.; Cucco, M.; Dalla Riva, G.V.; Giannini, E.G.; Piscaglia, F.; Rapaccini, G.; Di Marco, M.; Caturelli, E.; et al. Epidemiological Trends and Trajectories of MAFLD-Associated Hepatocellular Carcinoma 2002–2033: The ITA.LI.CA Database. Gut 2023, 72, 141–152. [Google Scholar] [CrossRef]
- Pennisi, G.; Enea, M.; Romero-Gomez, M.; Viganò, M.; Bugianesi, E.; Wong, V.W.-S.; Fracanzani, A.L.; Sebastiani, G.; Boursier, J.; Berzigotti, A.; et al. Liver-related and Extrahepatic Events in Patients with Non-alcoholic Fatty Liver Disease: A Retrospective Competing Risks Analysis. Aliment. Pharmacol. Ther. 2022, 55, 604–615. [Google Scholar] [CrossRef]
- Pennisi, G.; Pipitone, R.M.; Enea, M.; De Vincentis, A.; Battaglia, S.; Di Marco, V.; Di Martino, V.; Spatola, F.; Tavaglione, F.; Vespasiani-Gentilucci, U.; et al. A Genetic and Metabolic Staging System for Predicting the Outcome of Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2022, 6, 1032–1044. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Wattacheril, J. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Gastroenterol. Clin. N. Am. 2020, 49, 141–149. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Huby, T.; Gautier, E.L. Immune Cell-Mediated Features of Non-Alcoholic Steatohepatitis. Nat. Rev. Immunol. 2022, 22, 429–443. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive Immunity: An Emerging Player in the Progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-Aggressive CXCR6+ CD8 T Cells Cause Liver Immune Pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the Liver—From Homeostasis to Disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Peiseler, M.; Tacke, F. Inflammatory Mechanisms Underlying Nonalcoholic Steatohepatitis and the Transition to Hepatocellular Carcinoma. Cancers 2021, 13, 730. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Bogdanos, D.P.; Gao, B.; Gershwin, M.E. Liver Immunology. In Comprehensive Physiology; Terjung, R., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 567–598. ISBN 978-0-470-65071-4. [Google Scholar]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver Immunology and Its Role in Inflammation and Homeostasis. Cell Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Crispe, I.N. The Liver as a Lymphoid Organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef]
- Varol, C.; Yona, S.; Jung, S. Origins and Tissue-context-dependent Fates of Blood Monocytes. Immunol. Cell Biol. 2009, 87, 30–38. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 Signaling in Obese NAFLD. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef]
- Kanuri, G.; Ladurner, R.; Skibovskaya, J.; Spruss, A.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Expression of toll-like receptors 1–5 but not TLR 6-10 is elevated in livers of patients with non-alcoholic fatty liver disease. Liver Int. 2015, 35, 562–568. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, Metaflammation and Immunometabolic Disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Xu, G.-X.; Wei, S.; Yu, C.; Zhao, S.-Q.; Yang, W.-J.; Feng, Y.-H.; Pan, C.; Yang, K.-X.; Ma, Y. Activation of Kupffer Cells in NAFLD and NASH: Mechanisms and Therapeutic Interventions. Front. Cell Dev. Biol. 2023, 11, 1199519. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; Yao, Y.; Zhang, X.; Guan, Y.; Zheng, F. CD4+ T Cell Activation and Inflammation in NASH-Related Fibrosis. Front. Immunol. 2022, 13, 967410. [Google Scholar] [CrossRef]
- Matsuda, M.; Seki, E. The Liver Fibrosis Niche: Novel Insights into the Interplay between Fibrosis-Composing Mesenchymal Cells, Immune Cells, Endothelial Cells, and Extracellular Matrix. Food Chem. Toxicol. 2020, 143, 111556. [Google Scholar] [CrossRef]
- Ito, S.; Yukawa, T.; Uetake, S.; Yamauchi, M. Serum Intercellular Adhesion Molecule-1 in Patients with Nonalcoholic Steatohepatitis: Comparison with Alcoholic Hepatitis. Alcohol. Clin. Exp. Res. 2007, 31, S83–S87. [Google Scholar] [CrossRef]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver Inflammation Abrogates Immunological Tolerance Induced by Kupffer Cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune Surveillance by the Liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-Dependent and Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Dependent Manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and Immune Cells: An Important Regulatory Axis in the Tumor Microenvironment and Progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Bendelac, A.; Savage, P.B.; Teyton, L. The Biology of NKT Cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef]
- Kotas, M.E.; Lee, H.-Y.; Gillum, M.P.; Annicelli, C.; Guigni, B.A.; Shulman, G.I.; Medzhitov, R. Impact of CD1d Deficiency on Metabolism. PLoS ONE 2011, 6, e25478. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y. Role of NKT Cells in the Pathogenesis of NAFLD. Int. J. Hepatol. 2012, 2012, 850836. [Google Scholar] [CrossRef]
- Syn, W.-K.; Agboola, K.M.; Swiderska, M.; Michelotti, G.A.; Liaskou, E.; Pang, H.; Xie, G.; Philips, G.; Chan, I.S.; Karaca, G.F.; et al. NKT-Associated Hedgehog and Osteopontin Drive Fibrogenesis in Non-Alcoholic Fatty Liver Disease. Gut 2012, 61, 1323–1329. [Google Scholar] [CrossRef]
- Lombardi, R.; Piciotti, R.; Dongiovanni, P.; Meroni, M.; Fargion, S.; Fracanzani, A.L. PD-1/PD-L1 Immuno-Mediated Therapy in NAFLD: Advantages and Obstacles in the Treatment of Advanced Disease. Int. J. Mol. Sci. 2022, 23, 2707. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and Resolution of Inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef]
- Lu, J.G.; Iyasu, A.; French, B.; Tillman, B.; French, S.W. Overexpression of MHCII by Hepatocytes in Alcoholic Hepatitis (AH) Compared to Non-Alcoholic Steatohepatitis (NASH) and Normal Controls. Alcohol 2020, 84, 27–32. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Kubes, P. More Friend than Foe: The Emerging Role of Neutrophils in Tissue Repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as Protagonists and Targets in Chronic Inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Arelaki, S.; Koletsa, T.; Sinakos, E.; Papadopoulos, V.; Arvanitakis, K.; Skendros, P.; Akriviadis, E.; Ritis, K.; Germanidis, G.; Hytiroglou, P. Neutrophil Extracellular Traps Enriched with IL-1β and IL-17A Participate in the Hepatic Inflammatory Process of Patients with Non-Alcoholic Steatohepatitis. Virchows Arch. 2022, 481, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver Macrophages in Tissue Homeostasis and Disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Maretti-Mira, A.C.; Golden-Mason, L.; Salomon, M.P.; Kaplan, M.J.; Rosen, H.R. Cholesterol-Induced M4-Like Macrophages Recruit Neutrophils and Induce NETosis. Front. Immunol. 2021, 12, 671073. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, M.-J.; Cai, Y.; Wang, W.; Jiang, J.X.; Varga, Z.V.; Feng, D.; Pacher, P.; Kunos, G.; Torok, N.J.; et al. Neutrophil–Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 399–413. [Google Scholar] [CrossRef]
- Horst, A.K.; Tiegs, G.; Diehl, L. Contribution of Macrophage Efferocytosis to Liver Homeostasis and Disease. Front. Immunol. 2019, 10, 2670. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The Liver as an Immunological Organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic Inhibition of Inflammatory Monocyte Recruitment Reduces Steatohepatitis and Liver Fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef]
- De Silva, N.S.; Klein, U. Dynamics of B Cells in Germinal Centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte Responses to Oxidative Stress-Derived Antigens Contribute to the Evolution of Nonalcoholic Fatty Liver Disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Barrow, F.; Khan, S.; Fredrickson, G.; Wang, H.; Dietsche, K.; Parthiban, P.; Robert, S.; Kaiser, T.; Winer, S.; Herman, A.; et al. Microbiota-Driven Activation of Intrahepatic B Cells Aggravates NASH Through Innate and Adaptive Signaling. Hepatology 2021, 74, 704–722. [Google Scholar] [CrossRef] [PubMed]
- Litinskiy, M.B.; Nardelli, B.; Hilbert, D.M.; He, B.; Schaffer, A.; Casali, P.; Cerutti, A. DCs Induce CD40-Independent Immunoglobulin Class Switching through BLyS and APRIL. Nat. Immunol. 2002, 3, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Abe, M.; Tokumoto, Y.; Hirooka, M.; Furukawa, S.; Kumagi, T.; Hamada, M.; Kawasaki, K.; Tada, F.; Ueda, T.; et al. B Cell-Activating Factor Is Associated with the Histological Severity of Nonalcoholic Fatty Liver Disease. Hepatol. Int. 2013, 7, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Z.; Leung, M.W.; He, X.; Zhang, W.; Yang, G.; Leung, P.S.; Eric Gershwin, M. Adaptive Immunity in the Liver. Cell. Mol. Immunol. 2016, 13, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Podda, M.; Gershwin, M.E. Old and Rising Stars in the Lymphoid Liver. Semin. Immunopathol. 2009, 31, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Immunology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Taubert, R.; Hardtke-Wolenski, M.; Noyan, F.; Wilms, A.; Baumann, A.K.; Schlue, J.; Olek, S.; Falk, C.S.; Manns, M.P.; Jaeckel, E. Intrahepatic Regulatory T Cells in Autoimmune Hepatitis Are Associated with Treatment Response and Depleted with Current Therapies. J. Hepatol. 2014, 61, 1106–1114. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive Immune Responses Triggered by Oxidative Stress Contribute to Hepatic Inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef]
- Murphy, K.M.; Stockinger, B. Effector T Cell Plasticity: Flexibility in the Face of Changing Circumstances. Nat. Immunol. 2010, 11, 674–680. [Google Scholar] [CrossRef]
- Hirsova, P.; Bamidele, A.O.; Wang, H.; Povero, D.; Revelo, X.S. Emerging Roles of T Cells in the Pathogenesis of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Front. Endocrinol. 2021, 12, 760860. [Google Scholar] [CrossRef]
- Luo, X.-Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.-K. IFN-γ Deficiency Attenuates Hepatic Inflammation and Fibrosis in a Steatohepatitis Model Induced by a Methionine- and Choline-Deficient High-Fat Diet. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, G891–G899. [Google Scholar] [CrossRef]
- Omenetti, S.; Bussi, C.; Metidji, A.; Iseppon, A.; Lee, S.; Tolaini, M.; Li, Y.; Kelly, G.; Chakravarty, P.; Shoaie, S.; et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 2019, 51, 77–89.e6. [Google Scholar] [CrossRef]
- Xu, R.; Tao, A.; Zhang, S.; Zhang, M. Neutralization of Interleukin-17 Attenuates High Fat Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. Acta Biochim. Biophys. Sin. 2013, 45, 726–733. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Ma, H.-Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 Signaling in Steatotic Hepatocytes and Macrophages Promotes Hepatocellular Carcinoma in Alcohol-Related Liver Disease. J. Hepatol. 2020, 72, 946–959. [Google Scholar] [CrossRef]
- Wedemeyer, H.; He, X.-S.; Nascimbeni, M.; Davis, A.R.; Greenberg, H.B.; Hoofnagle, J.H.; Liang, T.J.; Alter, H.; Rehermann, B. Impaired Effector Function of Hepatitis C Virus-Specific CD8+ T Cells in Chronic Hepatitis C Virus Infection1. J. Immunol. 2002, 169, 3447–3458. [Google Scholar] [CrossRef]
- Kaech, S.M.; Cui, W. Transcriptional Control of Effector and Memory CD8+ T Cell Differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Breuer, D.A.; Pacheco, M.C.; Washington, M.K.; Montgomery, S.A.; Hasty, A.H.; Kennedy, A.J. CD8+ T Cells Regulate Liver Injury in Obesity-Related Nonalcoholic Fatty Liver Disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, G211–G224. [Google Scholar] [CrossRef]
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I Interferon Responses Drive Intrahepatic T Cells to Promote Metabolic Syndrome. Sci. Immunol. 2017, 2, eaai7616. [Google Scholar] [CrossRef]
- Bhattacharjee, J.; Kirby, M.; Softic, S.; Miles, L.; Salazar-Gonzalez, R.-M.; Shivakumar, P.; Kohli, R. Hepatic Natural Killer T-Cell and CD8+ T-Cell Signatures in Mice with Nonalcoholic Steatohepatitis. Hepatol. Commun. 2017, 1, 299–310. [Google Scholar] [CrossRef]
- Hansel, C.; Erschfeld, S.; Baues, M.; Lammers, T.; Weiskirchen, R.; Trautwein, C.; Kroy, D.C.; Drescher, H.K. The Inhibitory T Cell Receptors PD1 and 2B4 Are Differentially Regulated on CD4 and CD8 T Cells in a Mouse Model of Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2019, 10, 244. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.; He, Q.; Yang, B.; Cao, J. Small Molecule Inhibitors Targeting the PD-1/PD-L1 Signaling Pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Arasanz, H.; Gato-Cañas, M.; Zuazo, M.; Ibañez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 Signal Transduction Pathways in T Cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The Common γ-Chain Cytokines IL-2, IL-7, IL-15, and IL-21 Induce the Expression of Programmed Death-1 and Its Ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial Activation Chemicals Synergize with Surface Receptor PD-1 Blockade for T Cell-Dependent Antitumor Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef]
- O’Sullivan, D.; Pearce, E.L. Targeting T Cell Metabolism for Therapy. Trends Immunol. 2015, 36, 71–80. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 Antigen on the Surface of Stimulated Mouse T and B Lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L. PD-1 Signaling in Primary T Cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. PD-1 Increases PTEN Phosphatase Activity While Decreasing PTEN Protein Stability by Inhibiting Casein Kinase 2. Mol. Cell. Biol. 2013, 33, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New Insights into Its Biological Function and Use in Tumor Immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic Self-Tolerance Maintained by Cd25+Cd4+Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte–Associated Antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current Landscape and Future of Dual Anti-CTLA4 and PD-1/PD-L1 Blockade Immunotherapy in Cancer; Lessons Learned from Clinical Trials with Melanoma and Non-Small Cell Lung Cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, A.N.; Sharpe, A.H. Studies Using Antigen-Presenting Cells Lacking Expression of Both B7-1 (CD80) and B7-2 (CD86) Show Distinct Requirements for B7 Molecules During Priming Versus Restimulation of Th2 But Not Th1 Cytokine Production1. J. Immunol. 1998, 161, 2762–2771. [Google Scholar] [CrossRef]
- Sawada, K.; Chung, H.; Softic, S.; Moreno-Fernandez, M.E.; Divanovic, S. The Bidirectional Immune Crosstalk in Metabolic Dysfunction-Associated Steatotic Liver Disease. Cell Metab. 2023, 35, 1852–1871. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7–CD28 Superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Chatzigeorgiou, A.; Chung, K.-J.; Garcia-Martin, R.; Alexaki, V.-I.; Klotzsche-von Ameln, A.; Phieler, J.; Sprott, D.; Kanczkowski, W.; Tzanavari, T.; Bdeir, M.; et al. Dual Role of B7 Costimulation in Obesity-Related Nonalcoholic Steatohepatitis and Metabolic Dysregulation. Hepatology 2014, 60, 1196–1210. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Meneghel, P.; Farinati, F.; Russo, F.P.; Pelizzaro, F.; Gambato, M. Efficacy of Immunotherapy in Hepatocellular Carcinoma: Does Liver Disease Etiology Have a Role? Dig. Liver Dis. 1590. [Google Scholar] [CrossRef]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells within B16 Melanoma Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Tao, L.; Song, C.; Huo, C.; Sun, Y.; Zhang, C.; Li, X.; Yu, S.; Sun, M.; Jin, B.; Zhang, Z.; et al. Anti-CD155 and anti-CD112 monoclonal antibodies conjugated to a fluorescent mesoporous silica nanosensor encapsulating rhodamine 6G and fluorescein for sensitive detection of liver cancer cells. Analyst 2016, 141, 4933–4940. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a Novel Checkpoint for Human T Cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.; Lanier, L.L.; Shibuya, A. Functional Characterization of DNAM-1 (CD226) Interaction with Its Ligands PVR (CD155) and Nectin-2 (PRR-2/CD112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 Impair Tumor Antigen–Specific CD8+ T Cells in Melanoma Patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting Edge: TIGIT Has T Cell-Intrinsic Inhibitory Functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [PubMed]
- Ramsbottom, K.M.; Hawkins, E.D.; Shimoni, R.; McGrath, M.; Chan, C.J.; Russell, S.M.; Smyth, M.J.; Oliaro, J. Cutting Edge: DNAX Accessory Molecule 1–Deficient CD8+ T Cells Display Immunological Synapse Defects That Impair Antitumor Immunity. J. Immunol. 2014, 192, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of Obesity-Associated Insulin Resistance through Immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagai, R. Adipose Tissue Inflammation in Obesity and Metabolic Syndrome. Discov. Med. 2009, 8, 55–60. [Google Scholar] [PubMed]
- Deng, J.; Liu, S.; Zou, L.; Xu, C.; Geng, B.; Xu, G. Lipolysis Response to Endoplasmic Reticulum Stress in Adipose Cells *. J. Biol. Chem. 2012, 287, 6240–6249. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Nikolajczyk, B.S. Tissue Immune Cells Fuel Obesity-Associated Inflammation in Adipose Tissue and Beyond. Front. Immunol. 2019, 10, 1587. [Google Scholar] [CrossRef]
- Eljaafari, A.; Pestel, J.; Le Magueresse-Battistoni, B.; Chanon, S.; Watson, J.; Robert, M.; Disse, E.; Vidal, H. Adipose-Tissue-Derived Mesenchymal Stem Cells Mediate PD-L1 Overexpression in the White Adipose Tissue of Obese Individuals, Resulting in T Cell Dysfunction. Cells 2021, 10, 2645. [Google Scholar] [CrossRef]
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity Accelerates T Cell Senescence in Murine Visceral Adipose Tissue. J. Clin. Investig. 2016, 126, 4626–4639. [Google Scholar] [CrossRef]
- Oldenhove, G.; Boucquey, E.; Taquin, A.; Acolty, V.; Bonetti, L.; Ryffel, B.; Le Bert, M.; Englebert, K.; Boon, L.; Moser, M. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep. 2018; 25, 2053–2060.e4. [Google Scholar] [CrossRef]
- Schwartz, C.; Schmidt, V.; Deinzer, A.; Hawerkamp, H.C.; Hams, E.; Bayerlein, J.; Röger, O.; Bailer, M.; Krautz, C.; El Gendy, A.; et al. Innate PD-L1 Limits T Cell–Mediated Adipose Tissue Inflammation and Ameliorates Diet-Induced Obesity. Sci. Transl. Med. 2022, 14, eabj6879. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef]
- Nov, O.; Shapiro, H.; Ovadia, H.; Tarnovscki, T.; Dvir, I.; Shemesh, E.; Kovsan, J.; Shelef, I.; Carmi, Y.; Voronov, E.; et al. Interleukin-1β Regulates Fat-Liver Crosstalk in Obesity by Auto-Paracrine Modulation of Adipose Tissue Inflammation and Expandability. PLoS ONE 2013, 8, e53626. [Google Scholar] [CrossRef] [PubMed]
- Pedoeem, A.; Azoulay-Alfaguter, I.; Strazza, M.; Silverman, G.J.; Mor, A. Programmed Death-1 Pathway in Cancer and Autoimmunity. Clin. Immunol. 2014, 153, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jin, Q.; Nie, S.; Jia, S.; Li, Y.; Li, X.; Guo, F. Unlike PD-L1, PD-1 Is Downregulated on Partial Immune Cells in Type 2 Diabetes. J. Diabetes Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, R.; Haseda, F.; Tsutsumi, C.; Hiromine, Y.; Noso, S.; Kawabata, Y.; Mitsui, S.; Terasaki, J.; Ikegami, H.; Imagawa, A.; et al. Low Programmed Cell Death-1 (PD-1) Expression in Peripheral CD4+ T Cells in Japanese Patients with Autoimmune Type 1 Diabetes. Clin. Exp. Immunol. 2015, 180, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Liu, F.; Chen, K.; Tang, L.; Liu, L.; Zhang, K.; Yu, C.; Bian, G.; Guo, H.; Zheng, J.; et al. Programmed Death 1 Deficiency Induces the Polarization of Macrophages/Microglia to the M1 Phenotype After Spinal Cord Injury in Mice. Neurotherapeutics 2014, 11, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Piątkiewicz, P.; Bernat-Karpińska, M.; Miłek, T.; Rabijewski, M.; Rosiak, E. NK Cell Count and Glucotransporter 4 (GLUT4) Expression in Subjects with Type 2 Diabetes and Colon Cancer. Diabetol. Metab. Syndr. 2016, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired Mitochondrial Oxidative Phosphorylation Limits the Self-Renewal of T Cells Exposed to Persistent Antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef]
- Hashemi, M.; Karami, S.; Sarabandi, S.; Moazeni-Roodi, A.; Małecki, A.; Ghavami, S.; Wiechec, E. Association between PD-1 and PD-L1 Polymorphisms and the Risk of Cancer: A Meta-Analysis of Case-Control Studies. Cancers 2019, 11, 1150. [Google Scholar] [CrossRef] [PubMed]
- Eldafashi, N.; Darlay, R.; Shukla, R.; McCain, M.V.; Watson, R.; Liu, Y.L.; McStraw, N.; Fathy, M.; Fawzy, M.A.; Zaki, M.Y.W.; et al. A PDCD1 Role in the Genetic Predisposition to NAFLD-HCC? Cancers 2021, 13, 1412. [Google Scholar] [CrossRef] [PubMed]
- Pipitone, R.M.; Malvestiti, F.; Pennisi, G.; Jamialahmadi, O.; Dongiovanni, P.; Bertolazzi, G.; Pihlajamäki, J.; Yki-Järvinen, H.; Vespasiani-Gentilucci, U.; Tavaglione, F.; et al. Programmed Cell Death 1 Genetic Variant and Liver Damage in Nonalcoholic Fatty Liver Disease. Liver Int. 2023, 43, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Schöniger, S.; Jasani, B. The PD-1/PD-L1 Pathway: A Perspective on Comparative Immuno-Oncology. Animals 2022, 12, 2661. [Google Scholar] [CrossRef]
- Mao, T.; Yang, R.; Luo, Y.; He, K. Crucial Role of T Cells in NAFLD-Related Disease: A Review and Prospect. Front. Endocrinol. 2022, 13, 1051076. [Google Scholar] [CrossRef]
- Pennisi, G.; Celsa, C.; Spatola, F.; Dallio, M.; Federico, A.; Petta, S. Pharmacological Therapy of Non-Alcoholic Fatty Liver Disease: What Drugs Are Available Now and Future Perspectives. Int. J. Environ. Res. Public Health 2019, 16, 4334. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic Macrophages in Liver Homeostasis and Diseases-Diversity, Plasticity and Therapeutic Opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Raeman, R.; Anania, F.A. Therapy for Steatohepatitis: Do Macrophages Hold the Clue? Hepatology 2018, 67, 1204–1206. [Google Scholar] [CrossRef]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.-S.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential Expression and Activation of a Family of Murine Peroxisome Proliferator-Activated Receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic Acid for the Treatment of Non-Alcoholic Steatohepatitis: Interim Analysis from a Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Lucas, K.J.; Francque, S.; Abdelmalek, M.F.; Sanyal, A.J.; Ratziu, V.; Gadano, A.C.; Rinella, M.; Charlton, M.; Loomba, R.; et al. Tropifexor plus Cenicriviroc Combination versus Monotherapy in Nonalcoholic Steatohepatitis: Results from the Phase 2b TANDEM Study. Hepatology 2023, 78, 1223–1239. [Google Scholar] [CrossRef]
- Ilan, Y.; Shailubhai, K.; Sanyal, A. Immunotherapy with Oral Administration of Humanized Anti-CD3 Monoclonal Antibody: A Novel Gut-Immune System-Based Therapy for Metaflammation and NASH. Clin. Exp. Immunol. 2018, 193, 275–283. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pipitone, R.M.; Lupo, G.; Zito, R.; Javed, A.; Petta, S.; Pennisi, G.; Grimaudo, S. The PD-1/PD-L1 Axis in the Biology of MASLD. Int. J. Mol. Sci. 2024, 25, 3671. https://doi.org/10.3390/ijms25073671
Pipitone RM, Lupo G, Zito R, Javed A, Petta S, Pennisi G, Grimaudo S. The PD-1/PD-L1 Axis in the Biology of MASLD. International Journal of Molecular Sciences. 2024; 25(7):3671. https://doi.org/10.3390/ijms25073671
Chicago/Turabian StylePipitone, Rosaria Maria, Giulia Lupo, Rossella Zito, Ayesha Javed, Salvatore Petta, Grazia Pennisi, and Stefania Grimaudo. 2024. "The PD-1/PD-L1 Axis in the Biology of MASLD" International Journal of Molecular Sciences 25, no. 7: 3671. https://doi.org/10.3390/ijms25073671
APA StylePipitone, R. M., Lupo, G., Zito, R., Javed, A., Petta, S., Pennisi, G., & Grimaudo, S. (2024). The PD-1/PD-L1 Axis in the Biology of MASLD. International Journal of Molecular Sciences, 25(7), 3671. https://doi.org/10.3390/ijms25073671