
Chemokine Fractalkine and Non-Obstructive Coronary Artery Disease—Is There a Link?

 ,
, 
Abstract
:1. Introduction
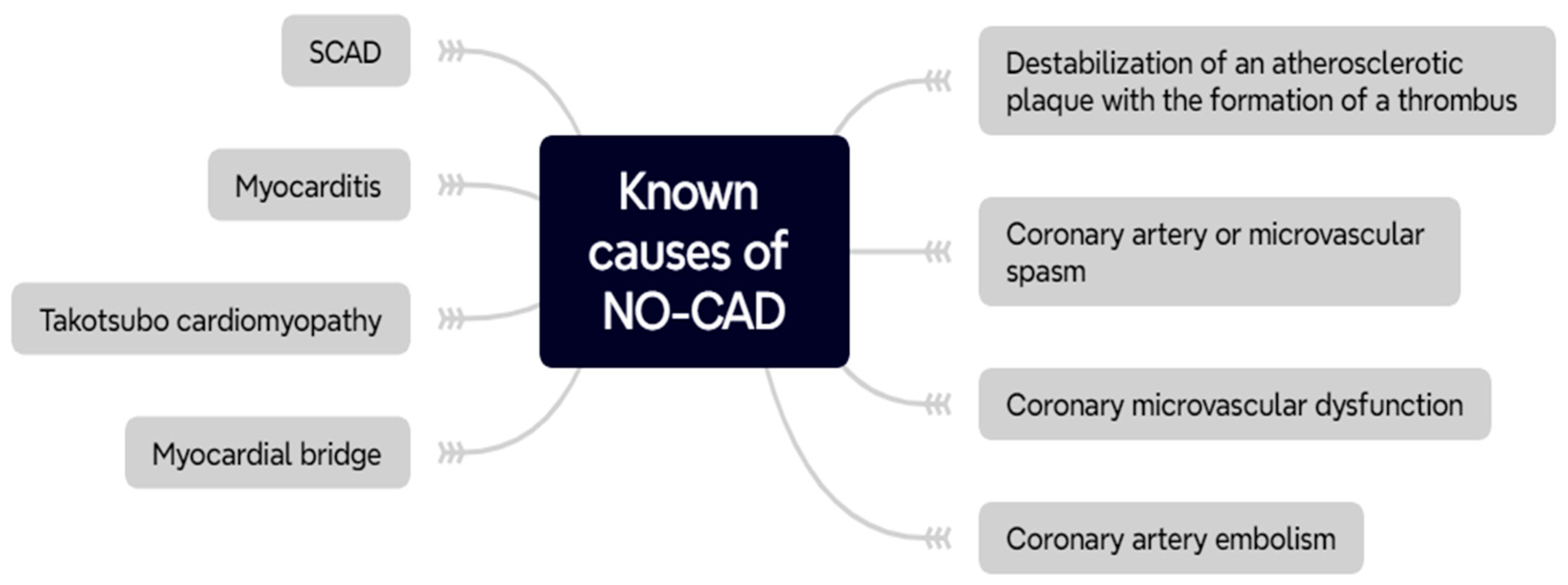
2. Non-Obstructive Coronary Artery Disease
2.1. Prevalence of NO-CAD
2.2. Pathophysiology of NO-CAD
2.2.1. Coronary Microvascular Dysfunction
2.2.2. Atherosclerosis and Plaque Destabilization
2.2.3. Inflammation
3. An Overview of Fractalkine
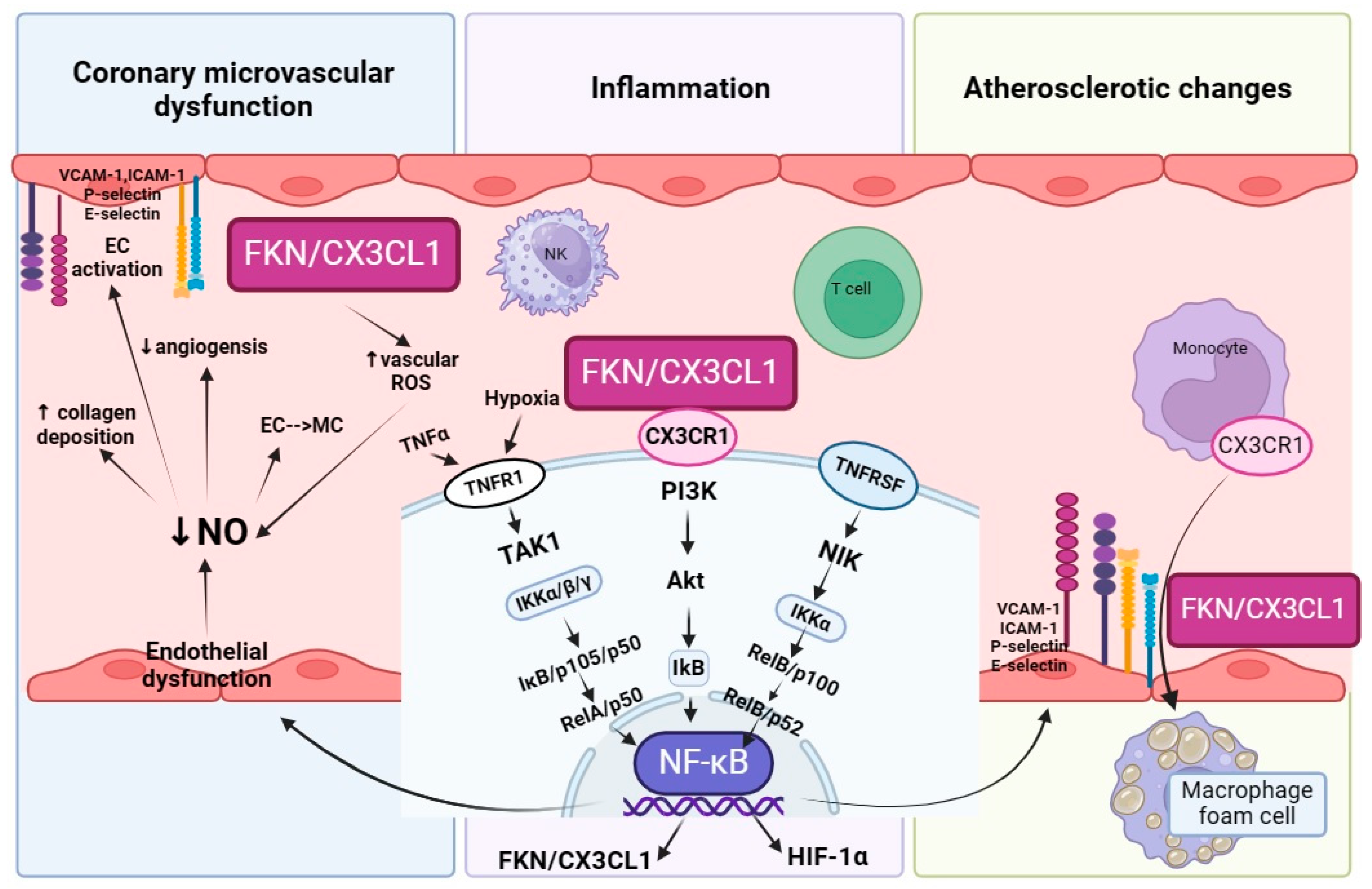
4. The Link between Fractalkine and NO-CAD
4.1. Fractalkine—A Key Player Which Promotes Inflammation
4.2. Fractalkine Underlies the Onset of Atherogenesis and Further Enhances This State
4.3. Fractalkine Promotes Coronary Microvascular Dysfunction
4.4. Future Implications for Fractalkine Role in NO-CAD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Shaw, L.J.; Shaw, R.E.; Bairey Merz, C.N.; Brindis, R.G.; Klein, L.W.; Nallamothu, B.; Douglas, P.S.; Krone, R.J.; McKay, C.R.; Block, P.C.; et al. Impact of Ethnicity and Gender Differences on Angiographic Coronary Artery Disease Prevalence and In-Hospital Mortality in the American College of Cardiology–National Cardiovascular Data Registry. Circulation 2008, 117, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-González, I. The Epidemiology of Coronary Heart Disease. Rev. Española De Cardiol. (Engl. Ed.) 2014, 67, 139–144. [Google Scholar] [CrossRef]
- Makarović, Z. Nonobstructive Coronary Artery Disease–Clinical Relevance, Diagnosis, Management and Proposal of New Pathophysiological Classification. Acta Clin. Croat. 2018, 57, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Pepine, C.J. ANOCA/INOCA/MINOCA: Open artery ischemia. Am. Heart J. Plus Cardiol. Res. Pract. 2023, 26, 100260. [Google Scholar] [CrossRef]
- Ford, T.J.; Yii, E.; Sidik, N.; Good, R.; Rocchiccioli, P.; McEntegart, M.; Watkins, S.; Eteiba, H.; Shaukat, A.; Lindsay, M.; et al. Ischemia and No Obstructive Coronary Artery Disease. Circ. Cardiovasc. Interv. 2019, 12, e008126. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.J.; Merz, C.N.B.; Pepine, C.J.; Reis, S.E.; Bittner, V.; Kip, K.E.; Kelsey, S.F.; Olson, M.; Johnson, B.D.; Mankad, S.; et al. The Economic Burden of Angina in Women With Suspected Ischemic Heart Disease. Circulation 2006, 114, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, L.; Hvelplund, A.; Abildstrom, S.Z.; Pedersen, F.; Galatius, S.; Madsen, J.K.; Jørgensen, E.; Kelbaek, H.; Prescott, E. Stable angina pectoris with no obstructive coronary artery disease is associated with increased risks of major adverse cardiovascular events. Eur. Heart J. 2012, 33, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Herscovici, R.; Sedlak, T.; Wei, J.; Pepine, C.J.; Handberg, E.; Bairey Merz, C.N. Ischemia and No Obstructive Coronary Artery Disease (INOCA): What Is the Risk? J. Am. Heart Assoc. 2018, 7, e008868. [Google Scholar] [CrossRef] [PubMed]
- Gehrie, E.R.; Reynolds, H.R.; Chen, A.Y.; Neelon, B.H.; Roe, M.T.; Gibler, W.B.; Ohman, E.M.; Newby, L.K.; Peterson, E.D.; Hochman, J.S. Characterization and outcomes of women and men with non–ST-segment elevation myocardial infarction and nonobstructive coronary artery disease: Results from the Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes with Early Implementation of the ACC/AHA Guidelines (CRUSADE) Quality Improvement Initiative. Am. Heart J. 2009, 158, 688–694. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.M.; Aa Fox, K.; White, J.A.; Giugliano, R.P.; Tricoci, P.; Reynolds, H.R.; Hochman, J.S.; Gibson, C.M.; Théroux, P.; Harrington, R.A.; et al. Outcomes among non-ST-segment elevation acute coronary syndromes patients with no angiographically obstructive coronary artery disease: Observations from 37,101 patients. Eur. Heart J. Acute Cardiovasc. Care 2014, 3, 37–45. [Google Scholar] [CrossRef]
- Bugiardini, R.; Merz, C.N.B. Angina With “Normal” Coronary Arteries: A Changing Philosophy. JAMA 2005, 293, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Herrmann, H.C.; Murphy, S.A.; Demopoulos, L.A.; DiBattiste, P.M.; Cannon, C.P.; Braunwald, E. Benefit of an Early Invasive Management Strategy in Women With Acute Coronary Syndromes. JAMA 2002, 288, 3124–3129. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.D.; Pepine, C.J. Gender Differences in the Treatment for Acute Myocardial Infarction. Circulation 2007, 115, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Fung, M.; Liang, Z.; Butalia, S.; Anderson, T.J. Temporal Trends of the Prevalence of Angina With No Obstructive Coronary Artery Disease (ANOCA). Can. J. Cardiol. 2023, 39, 63–70. [Google Scholar] [CrossRef]
- Abdu, F.A.; Mohammed, A.Q.; Liu, L.; Xu, Y.; Che, W. Myocardial Infarction with Nonobstructive Coronary Arteries (MINOCA): A Review of the Current Position. Cardiology 2020, 145, 543–552. [Google Scholar] [CrossRef]
- Rakowski, T.; De Luca, G.; Siudak, Z.; Plens, K.; Dziewierz, A.; Kleczyński, P.; Tokarek, T.; Węgiel, M.; Sadowski, M.; Dudek, D. Characteristics of patients presenting with myocardial infarction with non-obstructive coronary arteries (MINOCA) in Poland: Data from the ORPKI national registry. J. Thromb. Thrombolysis 2019, 47, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Smilowitz, N.R.; Mahajan, A.M.; Roe, M.T.; Hellkamp, A.S.; Chiswell, K.; Gulati, M.; Reynolds, H.R. Mortality of Myocardial Infarction by Sex, Age, and Obstructive Coronary Artery Disease Status in the ACTION Registry-GWTG (Acute Coronary Treatment and Intervention Outcomes Network Registry-Get with the Guidelines). Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003443. [Google Scholar] [CrossRef]
- Kühl, U.; Pauschinger, M.; Bock, T.; Klingel, K.; Schwimmbeck, C.P.L.; Seeberg, B.; Krautwurm, L.; Poller, W.; Schultheiss, H.-P.; Kandolf, R.; et al. Parvovirus B19 Infection Mimicking Acute Myocardial Infarction. Circulation 2003, 108, 945–950. [Google Scholar] [CrossRef]
- Lindahl, B.; Baron, T.; Albertucci, M.; Prati, F. Myocardial infarction with non-obstructive coronary artery disease. EuroIntervention 2021, 17, e875–e887. [Google Scholar] [CrossRef]
- Rehan, R.; Yong, A.; Ng, M.; Weaver, J.; Puranik, R. Coronary microvascular dysfunction: A review of recent progress and clinical implications. Front. Cardiovasc. Med. 2023, 10, 1111721. [Google Scholar] [CrossRef]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical Vasoconstriction Induced by Acetylcholine in Atherosclerotic Coronary Arteries. N. Engl. J. Med. 2009, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Allbritton-King, J.D.; García-Cardeña, G. Endothelial cell dysfunction in cardiac disease: Driver or consequence? Front. Cell Dev. Biol. 2023, 11, 1278166. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. Nitric oxide and the vascular endothelium. Handb. Exp. Pharmacol. 2006, 176, 213–254. [Google Scholar] [CrossRef]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef]
- Juni, R.P.; Kuster, D.W.D.; Goebel, M.; Helmes, M.; Musters, R.J.P.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.V.; Harrison, D.G.; Olbrych, M.T.; Alexander, R.W.; Medford, R.M. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 9114–9119. [Google Scholar] [CrossRef]
- Riad, A.; Westermann, D.; Linthout, S.; Van Mohr, Z.; Uyulmaz, S.; Becher, P.M.; Rütten, H.; Wohlfart, P.; Peters, H.; Schultheiss, H.-P.; et al. Enhancement of endothelial nitric oxide synthase production reverses vascular dysfunction and inflammation in the hindlimbs of a rat model of diabetes. Diabetologia 2008, 51, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Nagel, T.; Resnick, N.; Atkinson, W.J.; Dewey, C.F.; Gimbrone, M.A. Shear stress selectively upregulates intercellular adhesion molecule-1 expression in cultured human vascular endothelial cells. J. Clin. Investig. 1994, 94, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Shatanawi, A.; Romero, M.J.; Iddings, J.A.; Chandra, S.; Umapathy, N.S.; Verin, A.D.; Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; et al. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am. J. Physiol. Cell Physiol. 2011, 300, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Ushio-Fukai, M.; Lassègue, B.; Alexander, R.W. Angiotensin II Signaling in Vascular Smooth Muscle. Hypertension 1997, 29, 366–373. [Google Scholar] [CrossRef]
- Touyz, R.M. The role of angiotensin II in regulating vascular structural and functional changes in hypertension. Curr. Hypertens. Rep. 2003, 5, 155–164. [Google Scholar] [CrossRef]
- Scalera, F. Intracellular glutathione and lipid peroxide availability and the secretion of vasoactive substances by human umbilical vein endothelial cells after incubation with TNF-α. Eur. J. Clin. Investig. 2003, 33, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting Actions of Endothelin ETA and ETB Receptors in Cardiovascular Disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef]
- Marasciulo, F.L.; Montagnani, M.; Potenza, M.A. Endothelin-1: The Yin and Yang on Vascular Function. Curr. Med. Chem. 2006, 13, 1655–1665. [Google Scholar] [CrossRef]
- Camici, P.G.; Crea, F. Coronary Microvascular Dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Taqueti, V.R.; Di Carli, M.F. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options. J. Am. Coll. Cardiol. 2018, 72, 2625–2641. [Google Scholar] [CrossRef] [PubMed]
- Ashokprabhu, N.D.; Quesada, O.; Alvarez, Y.R.; Henry, T.D. INOCA/ANOCA: Mechanisms and novel treatments. Am. Heart J. Plus Cardiol. Res. Pract. 2023, 30, 100302. [Google Scholar] [CrossRef]
- Dayanikli, F.; Grambow, D.; Muzik, O.; Mosca, L.; Rubenfire, M.; Schwaiger, M. Early detection of abnormal coronary flow reserve in asymptomatic men at high risk for coronary artery disease using positron emission tomography. Circulation 1994, 90, 808–817. [Google Scholar] [CrossRef]
- Gould, K.L.; Martucci, J.P.; Goldberg, D.I.; Hess, M.J.; Edens, R.P.; Latifi, R.; Dudrick, S.J. Short-term cholesterol lowering decreases size and severity of perfusion abnormalities by positron emission tomography after dipyridamole in patients with coronary artery disease. A potential noninvasive marker of healing coronary endothelium. Circulation 1994, 89, 1530–1538. [Google Scholar] [CrossRef]
- Czernin, J.; Barnard, R.J.; Sun, K.T.; Krivokapich, J.; Nitzsche, E.; Dorsey, D.; Phelps, M.E.; Schelbert, H.R. Effect of Short-term Cardiovascular Conditioning and Low-Fat Diet on Myocardial Blood Flow and Flow Reserve. Circulation 1995, 92, 197–204. [Google Scholar] [CrossRef]
- Nitenberg, A.; Valensi, P.; Sachs, R.; Dali, M.; Aptecar, E.; Attali, J.R. Impairment of Coronary Vascular Reserve and ACh-Induced Coronary Vasodilation in Diabetic Patients With Angiographically Normal Coronary Arteries and Normal Left Ventricular Systolic Function. Diabetes 1993, 42, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, O.P.; Nuutila, P.; Raitakari, O.T.; Rönnemaa, T.; Koskinen, P.J.; Iida, H.; Lehtimäki, T.J.; Laine, H.K.; Takala, T.; Viikari, J.S.A.; et al. Coronary Flow Reserve Is Reduced in Young Men with IDDM. Diabetes 1998, 47, 248–254. [Google Scholar] [CrossRef]
- Brush, J.E., Jr.; Cannon, R.O.I.; Schenke, W.H.; Bonow, R.O.; Leon, M.B.; Maron, B.J.; Epstein, S.E. Angina Due to Coronary Microvascular Disease in Hypertensive Patients without Left Ventricular Hypertrophy. N. Engl. J. Med. 2010, 319, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Opherk, D.; Mall, G.; Zebe, H.; Schwarz, F.; Weihe, E.; Manthey, J.; Kübler, W. Reduction of coronary reserve: A mechanism for angina pectoris in patients with arterial hypertension and normal coronary arteries. Circulation 1984, 69, 1–7. [Google Scholar] [CrossRef]
- Camici, P.; Chiriatti, G.; Lorenzoni, R.; Bellina, R.C.; Gistri, R.; Italiani, G.; Parodi, O.; Salvadori, P.A.; Nista, N.; Papi, L.; et al. Coronary vasodilation is impaired in both hypertrophied and nonhypertrophied myocardium of patients with hypertrophic cardiomyopathy: A study with nitrogen-13 ammonia and positron emission tomography. J. Am. Coll. Cardiol. 1991, 17, 879–886. [Google Scholar] [CrossRef]
- Choudhury, L.; Elliott, P.; Rimoldi, O.; Ryan, M.; Lammertsma, A.A.; Boyd, H.; McKenna, W.; Camici, P. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res. Cardiol. 1999, 94, 49–59. [Google Scholar] [CrossRef]
- Marcus, M.L.; Harrison, D.G.; Chilian, W.M.; Koyanagi, S.; Inou, T.; Tomanek, R.J.; Martins, J.B.; Eastham, C.L.; Hiratzka, L.F. Alterations in the coronary circulation in hypertrophied ventricles. Circulation 1987, 75, I19–I25. [Google Scholar] [PubMed]
- Choudhury, L.; Rosen, S.D.; Patel, D.; Nihoyannopoulos, P.; Camici, P.G. Coronary vasodilator reserve in primary and secondary left ventricular hypertrophy: A study with positron emission tomography. Eur. Heart J. 1997, 18, 108–116. [Google Scholar] [CrossRef]
- Kaufmann, P.A.; Gnecchi-Ruscone, T.; Di Terlizzi, M.; Schäfers, K.P.; Lüscher, T.F.; Camici, P.G. Coronary Heart Disease in Smokers. Circulation 2000, 102, 1233–1238. [Google Scholar] [CrossRef]
- Khuddus, M.A.; Pepine, C.J.; Handberg, E.M.; Bairey Merz, C.N.; Sopko, G.; Bavry, A.A.; Denardo, S.J.; McGorray, S.P.; Smith, K.M.; Sharaf, B.L.; et al. An Intravascular Ultrasound Analysis in Women Experiencing Chest Pain in the Absence of Obstructive Coronary Artery Disease: A Substudy from the National Heart, Lung and Blood Institute–Sponsored Women’s Ischemia Syndrome Evaluation (WISE). J. Interv. Cardiol. 2010, 23, 511–519. [Google Scholar] [CrossRef]
- Krieglstein, C.F.; Granger, D.N. Adhesion molecules and their role in vascular disease. Am. J. Hypertens. 2001, 14, S44–S54. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Kolattukudy, P.E. Role of MCP-1 in cardiovascular disease: Molecular mechanisms and clinical implications. Clin. Sci. 2009, 117, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Berliner, J.; Leitinger, N.; Watson, A.; Huber, J.; Fogelman, A.; Navab, M. Oxidized Lipids in Atherogenesis: Formation, Destruction and Action. Thromb. Haemost. 1997, 78, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.H.; Tripathi, J.; Mishra, N.K.; Cai, Y.; Tripathi, S.; Wang, X.P.; Imes, S.; Fishbein, M.C.; Clinton, S.K.; Libby, P.; et al. Role of macrophage colony-stimulating factor in atherosclerosis: Studies of osteopetrotic mice. Am. J. Pathol. 1997, 150, 1687–1699. [Google Scholar] [PubMed]
- Hansson, G.K. Immune Mechanisms in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2001, 21, 1876–1890. [Google Scholar] [CrossRef] [PubMed]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Libby, P. Current Concepts of the Pathogenesis of the Acute Coronary Syndromes. Circulation 2001, 104, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, D.; Capodanno, D.; Dangas, G.; Tamburino, C. Spontaneous coronary artery dissection. Int. J. Cardiol. 2014, 175, 8–20. [Google Scholar] [CrossRef]
- Lebrun, S.; Bond, R.M. Spontaneous coronary artery dissection (SCAD): The underdiagnosed cardiac condition that plagues women. Trends Cardiovasc. Med. 2018, 28, 340–345. [Google Scholar] [CrossRef]
- Libby, P.; Simon, D.I. Inflammation and Thrombosis. Circulation 2001, 103, 1718–1720. [Google Scholar] [CrossRef]
- Zhai, C.; Fan, H.; Zhu, Y.; Chen, Y.; Shen, L. Coronary functional assessment in non-obstructive coronary artery disease: Present situation and future direction. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Bhatt, D.L.; Pradhan, A.D.; Glynn, R.J.; MacFadyen, J.G.; Nissen, S.E. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: A collaborative analysis of three randomised trials. Lancet 2023, 401, 1293–1301. [Google Scholar] [CrossRef]
- Daniel, M.; Ekenbäck, C.; Agewall, S.; Brolin, E.B.; Caidahl, K.; Cederlund, K.; Collste, O.; Eurenius, L.; Frick, M.; Younis-Hassan, S.; et al. Risk Factors and Markers for Acute Myocardial Infarction with Angiographically Normal Coronary Arteries. Am. J. Cardiol. 2015, 116, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Hjort, M.; Eggers, K.M.; Lindhagen, L.; Agewall, S.; Brolin, E.B.; Collste, O.; Daniel, M.; Ekenbäck, C.; Frick, M.; Henareh, L.; et al. Increased Inflammatory Activity in Patients 3 Months after Myocardial Infarction with Nonobstructive Coronary Arteries. Clin. Chem. 2019, 65, 1023–1030. [Google Scholar] [CrossRef]
- Pober, J.S.; Cotran, R.S. Cytokines and endothelial cell biology. Physiol. Rev. 1990, 70, 427–451. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Citation: Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Sohn, R.H.; Deming, C.B.; Johns, D.C.; Champion, H.C.; Bian, C.; Gardner, K.; Rade, J.J. Regulation of endothelial thrombomodulin expression by inflammatory cytokines is mediated by activation of nuclear factor-kappa B. Blood 2005, 105, 3910–3917. [Google Scholar] [CrossRef]
- Dhanesha, N.; Prakash, P.; Doddapattar, P.; Khanna, I.; Pollpeter, M.J.; Nayak, M.K.; Staber, J.M.; Chauhan, A.K. Endothelial cell-derived von willebrand factor is the major determinant that mediates von willebrand factor-dependent acute Ischemic Stroke by promoting postischemic thrombo-inflammation. Arter. Thromb. Vasc. Biol. 2016, 36, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Spronk, H.M.H.; ten Cate, H. The Hemostatic System as a Modulator of Atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.-Y. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, e5. [Google Scholar] [CrossRef] [PubMed]
- Goda, S.; Imai, T.; Yoshie, O.; Yoneda, O.; Inoue, H.; Nagano, Y.; Okazaki, T.; Imai, H.; Bloom, E.T.; Domae, N.; et al. CX3C-Chemokine, Fractalkine-Enhanced Adhesion of THP-1 Cells to Endothelial Cells Through Integrin-Dependent and -Independent Mechanisms. J. Immunol. 2000, 164, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and Molecular Characterization of Fractalkine Receptor CX3CR1, which Mediates Both Leukocyte Migration and Adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Skoda, M.; Stangret, A.; Szukiewicz, D. Fractalkine and placental growth factor: A duet of inflammation and angiogenesis in cardiovascular disorders. Cytokine Growth Factor Rev. 2018, 39, 116–123. [Google Scholar] [CrossRef]
- Umehara, H.; Bloom, E.T.; Okazaki, T.; Nagano, Y.; Yoshie, O.; Imai, T. Fractalkine in Vascular Biology. Arter. Thromb. Vasc. Biol. 2004, 24, 34–40. [Google Scholar] [CrossRef]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor Necrosis Factor-α-converting Enzyme (ADAM17) Mediates the Cleavage and Shedding of Fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Perla, R.P.; Bysani, S.; Dulin, N.O.; Liu, F.; Melby, P.C. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem. J. 2003, 373, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Umehara, H.; Bloom, E.; Okazaki, T.; Domae, N.; Imai, T. Fractalkine and vascular injury. Trends Immunol. 2001, 22, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Namkoong, S.; Kim, Y.-M.; Kim, C.-K.; Lee, H.; Ha, K.-S.; Chung, H.-T.; Kwon, Y.-G.; Kim, Y.-M. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H2836–H2846. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Ou, J.; Zhang, S.; Ming, Y. Crosstalk between the CX3CL1/CX3CR1 Axis and Inflammatory Signaling Pathways in Tissue Injury. Curr. Protein Pept. Sci. 2019, 20, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.C.; Wong, D.; McManus, B.M. Characterization of fractalkine (CX3CL1) and CX3CR1 in human coronary arteries with native atherosclerosis, diabetes mellitus, and transplant vascular disease. Cardiovasc. Pathol. 2002, 11, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Feng, X.; Cai, W.; Liu, T.; Liang, Z.; Sun, Y.; Yan, C.; Han, Y. Chemokine CX3CL1 and its receptor CX3CR1 are associated with human atherosclerotic lesion volnerability. Thromb. Res. 2015, 135, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Matthews, G.J.; Shah, R.Y.; McLaughlin, C.; Chen, J.; Wolman, M.; Master, S.R.; Chai, B.; Xie, D.; Rader, D.J.; et al. Serum Fractalkine (CX3CL1) and Cardiovascular Outcomes and Diabetes: Findings From the Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. 2015, 66, 266–273. [Google Scholar] [CrossRef]
- Loh, S.X.; Ekinci, Y.; Spray, L.; Jeyalan, V.; Olin, T.; Richardson, G.; Austin, D.; Alkhalil, M.; Spyridopoulos, I. Fractalkine Signalling (CX3CL1/CX3CR1 Axis) as an Emerging Target in Coronary Artery Disease. J. Clin. Med. 2023, 12, 4821. [Google Scholar] [CrossRef]
- Guzik, T.J.; Skiba, D.S.; Touyz, R.M.; Harrison, D.G. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res. 2017, 113, 1009–1023. [Google Scholar] [CrossRef]
- Schober, A.; Zernecke, A. Chemokines in vascular remodeling. Thromb. Haemost. 2007, 97, 730–737. [Google Scholar] [CrossRef]
- Zheng, C.; Xuan, W.; Chen, Z.; Zhang, R.; Huang, X.; Zhu, Y.; Ma, S.; Chen, K.; Chen, L.; He, M.; et al. CX3CL1 Worsens Cardiorenal Dysfunction and Serves as a Therapeutic Target of Canagliflozin for Cardiorenal Syndrome. Front. Pharmacol. 2022, 13, 848310. [Google Scholar] [CrossRef] [PubMed]
- Cormican, S.; Griffin, M.D. Fractalkine (CX3CL1) and Its Receptor CX3CR1: A Promising Therapeutic Target in Chronic Kidney Disease? Front. Immunol. 2021, 12, 664202. [Google Scholar] [CrossRef]
- Vancheri, F.; Longo, G.; Vancheri, S.; Henein, M. Coronary Microvascular Dysfunction. J. Clin. Med. 2020, 9, 2880. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. Microvascular rarefaction. Organogenesis 2010, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, E.; Mendelev, N.; Patschan, S.; Patschan, D.; Eskander, J.; Cohen-Gould, L.; Chander, P.; Goligorsky, M.S. Chronic NOS inhibition actuates endothelial-mesenchymal transformation. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H285–H294. [Google Scholar] [CrossRef] [PubMed]
- Flierl, U.; Bauersachs, J.; Schäfer, A. Modulation of platelet and monocyte function by the chemokine fractalkine (CX3CL1) in cardiovascular disease. Eur. J. Clin. Investig. 2015, 45, 624–633. [Google Scholar] [CrossRef]
- Schäfer, A.; Schulz, C.; Eigenthaler, M.; Fraccarollo, D.; Kobsar, A.; Gawaz, M.; Ertl, G.; Walter, U.; Bauersachs, J. Novel role of the membrane-bound chemokine fractalkine in platelet activation and adhesion. Blood 2004, 103, 407–412. [Google Scholar] [CrossRef]
- Meyer dos Santos, S.; Klinkhardt, U.; Scholich, K.; Nelson, K.; Monsefi, N.; Deckmyn, H.; Kuczka, K.; Zorn, A.; Harder, S. The CX3C chemokine fractalkine mediates platelet adhesion via the von Willebrand receptor glycoprotein Ib. Blood 2011, 117, 4999–5008. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Leukocyte–endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003, 24, 326–333. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Hegele, R.A. The fractalkine receptor CX3CR1 is a key mediator of atherogenesis. J. Clin. Investig. 2003, 111, 1118–1120. [Google Scholar] [CrossRef]
- Kawamura, A.; Miura, S.; Fujino, M.; Nishikawa, H.; Matsuo, Y.; Tanigawa, H.; Tomita, S.; Tsuchiya, Y.; Matsuo, K.; Saku, K. CXCR3 Chemokine Receptor-Plasma IP10 Interaction in Patients With Coronary Artery Disease. Circ. J. 2003, 67, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, P.; Haskell, C.A.; Charo, I.F. Decreased atherosclerosis in CX3CR1–/– mice reveals a role for fractalkine in atherogenesis. J. Clin. Investig. 2003, 111, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Volger, O.L.; Fledderus, J.O.; Kisters, N.; Fontijn, R.D.; Moerland, P.D.; Kuiper, J.; van Berkel, T.J.; Bijnens, A.-P.J.; Daemen, M.J.; Pannekoek, H.; et al. Distinctive Expression of Chemokines and Transforming Growth Factor-β Signaling in Human Arterial Endothelium during Atherosclerosis. Am. J. Pathol. 2007, 171, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Stolla, M.; Pelisek, J.; von Brühl, M.-L.; Schäfer, A.; Barocke, V.; Heider, P.; Lorenz, M.; Tirniceriu, A.; Steinhart, A.; Bauersachs, J.; et al. Fractalkine Is Expressed in Early and Advanced Atherosclerotic Lesions and Supports Monocyte Recruitment via CX3CR1. PLoS ONE 2012, 7, e43572. [Google Scholar] [CrossRef] [PubMed]
- Poupel, L.; Boissonnas, A.; Hermand, P.; Dorgham, K.; Guyon, E.; Auvynet, C.; Charles, F.S.; Lesnik, P.; Deterre, P.; Combadiere, C. Pharmacological Inhibition of the Chemokine Receptor, CX3CR1, Reduces Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2013, 33, 2297–2305. [Google Scholar] [CrossRef]
- Cheng, C.; Tempel, D.; van Haperen, R.; de Boer, H.C.; Segers, D.; Huisman, M.; van Zonneveld, A.J.; Leenen, P.J.; van der Steen, A.; Serruys, P.W.; et al. Shear stress–induced changes in atherosclerotic plaque composition are modulated by chemokines. J. Clin. Investig. 2007, 117, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Babendreyer, A.; Molls, L.; Dreymueller, D.; Uhlig, S.; Ludwig, A. Shear Stress Counteracts Endothelial CX3CL1 Induction and Monocytic Cell Adhesion. Mediat. Inflamm 2017, 2017, 1515389. [Google Scholar] [CrossRef] [PubMed]
- Ruze, A.; Zhao, Y.; Li, H.; Gulireba, X.; Li, J.; Lei, D.; Dai, H.; Wu, J.; Zhao, X.; Nie, Y. Low shear stress upregulates the expression of fractalkine through the activation of mitogen-activated protein kinases in endothelial cells. Blood Coagul. Fibrinolysis 2018, 29, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Schäfer, A.; Schulz, C.; Fraccarollo, D.; Tas, P.; Leutke, M.; Eigenthaler, M.; Seidl, S.; Heider, P.; Ertl, G.; Massberg, S.; et al. The CX3C Chemokine Fractalkine Induces Vascular Dysfunction by Generation of Superoxide Anions. Arter. Thromb. Vasc. Biol. 2007, 27, 55–62. [Google Scholar] [CrossRef]
- Tousoulis, D.; Daves, G.J.; Asimakopoulos, G.; Homaei, H.; Zouridakis, E.; Ahmed, N.; Kaski, J.C. Vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 serum level in patients with chest pain and normal coronary arteries (syndrome X). Clin. Cardiol. 2001, 24, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-F.; Tsao, Y.-T.; Hou, C.-H. Fractalkine/CX3CL1 induced intercellular adhesion molecule-1-dependent tumor metastasis through the CX3CR1/PI3K/Akt/NF-κB pathway in human osteosarcoma. Oncotarget 2017, 8, 54136–54148. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Hinkle, C.C.; Ferguson, J.F.; Mehta, N.N.; Li, M.; Qu, L.; Lu, Y.; Putt, M.E.; Ahima, R.S.; Reilly, M.P. Fractalkine Is a Novel Human Adipochemokine Associated with Type 2 Diabetes. Diabetes 2011, 60, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zheng, M.; Hou, H.; Fang, S.; Chen, L.; Yang, J.; Yao, W.; Zhang, Q.; Hei, Z. Cellular senescence in ischemia/reperfusion injury. Cell Death Discov. 2022, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech. Ageing Dev. 2021, 198, 111540. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Cell Biology of Atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef]
- Combadiere, C.; Potteaux, S.; Gao, J.-L.; Esposito, B.; Casanova, S.; Lee, E.J.; Debre, P.; Tedgui, A.; Murphy, P.M.; Mallat, Z.; et al. Decreased Atherosclerotic Lesion Formation in CX3CR1/Apolipoprotein E Double Knockout Mice. Circulation 2003, 107, 1009–1016. [Google Scholar] [CrossRef]
- Greaves, D.R.; Häkkinen, T.; Lucas, A.D.; Liddiard, K.; Jones, E.; Quinn, C.M.; Senaratne, J.; Green, F.R.; Tyson, K.; Boyle, J.; et al. Linked Chromosome 16q13 Chemokines, Macrophage-Derived Chemokine, Fractalkine, and Thymus- and Activation-Regulated Chemokine, Are Expressed in Human Atherosclerotic Lesions. Arter. Thromb. Vasc. Biol. 2001, 21, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, A.; Potashnikova, D.; Freeman, M.L.; Gontarenko, V.; Maytesyan, D.; Lederman, M.M.; Vasilieva, E.; Margolis, L. Driving T cells to human atherosclerotic plaques: CCL3/CCR5 and CX3CL1/CX3CR1 migration axes. Eur. J. Immunol. 2021, 51, 1857–1859. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Schäfer, A.; Stolla, M.; Kerstan, S.; Lorenz, M.; von Brühl, M.-L.; Schiemann, M.; Bauersachs, J.; Gloe, T.; Busch, D.H.; et al. Chemokine Fractalkine Mediates Leukocyte Recruitment to Inflammatory Endothelial Cells in Flowing Whole Blood. Circulation 2007, 116, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoaty, S.; Arthur, H.; Spyridopoulos, I.; Wagberg, M.; Fritsche Danielson, R.; Pernow, J.; Gabrielsen, A.; Olin, T. 5234KAND567, the first selective small molecule CX3CR1 antagonist in clinical development, mediates anti-inflammatory cardioprotective effects in rodent models of atherosclerosis and myocardial infarction. Eur. Heart J. 2019, 40, ehz746.0080. [Google Scholar] [CrossRef]
- Ikejima, H.; Imanishi, T.; Tsujioka, H.; Kashiwagi, M.; Kuroi, A.; Tanimoto, T.; Kitabata, H.; Ishibashi, K.; Komukai, K.; Takeshita, T.; et al. Upregulation of Fractalkine and Its Receptor, CX3CR1, is Associated With Coronary Plaque Rupture in Patients with Unstable Angina Pectoris. Circ. J. 2010, 74, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Njerve, I.U.; Solheim, S.; Lunde, K.; Hoffmann, P.; Arnesen, H.; Seljeflot, I. Fractalkine levels are elevated early after PCI-treated ST-elevation myocardial infarction; no influence of autologous bone marrow derived stem cell injection. Cytokine 2014, 69, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zhang, S.; Lu, H.; Hong, X.; Qian, J.; Sun, A.; Zou, Y.; Ge, J. Changes in fractalkine in patients with ST-elevation myocardial infarction. Coron. Artery Dis. 2015, 26, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Boag, S.E.; Das, R.; Shmeleva, E.V.; Bagnall, A.; Egred, M.; Howard, N.; Bennaceur, K.; Zaman, A.; Keavney, B.; Spyridopoulos, I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J. Clin. Investig. 2015, 125, 3063–3076. [Google Scholar] [CrossRef] [PubMed]
- Stangret, A.; Dykacz, W.; Jabłoński, K.; Wesołowska, A.; Klimczak-Tomaniak, D.; Kochman, J.; Tomaniak, M. The cytokine trio-visfatin, placental growth factor and fractalkine–and their role in myocardial infarction with non-obstructive coronary arteries (MINOCA). Cytokine Growth Factor Rev. 2023, 74, 76–85. [Google Scholar] [CrossRef]
- Yoneda, O.; Imai, T.; Goda, S.; Inoue, H.; Yamauchi, A.; Okazaki, T.; Imai, H.; Yoshie, O.; Bloom, E.T.; Domae, N.; et al. Fractalkine-Mediated Endothelial Cell Injury by NK Cells. J. Immunol. 2000, 164, 4055–4062. [Google Scholar] [CrossRef]
- Kumar, A.; Hung, O.Y.; Piccinelli, M.; Eshtehardi, P.; Corban, M.T.; Sternheim, D.; Yang, B.; Lefieux, A.; Molony, D.S.; Thompson, E.W.; et al. Low Coronary Wall Shear Stress Is Associated with Severe Endothelial Dysfunction in Patients with Nonobstructive Coronary Artery Disease. JACC Cardiovasc. Interv. 2018, 11, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Sara, J.D.; Zaromytidou, M.; Park, K.H.; Coskun, A.U.; Lerman, L.O.; Oikonomou, E.; Maynard, C.C.; Fotiadis, D.; Stefanou, K.; et al. Local Low Shear Stress and Endothelial Dysfunction in Patients With Nonobstructive Coronary Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Patil, S.; Rojas, M.; Fong, A.M.; Smyth, S.S.; Patel, D.D. CX3 CR1 Deficiency Confers Protection From Intimal Hyperplasia After Arterial Injury. Arter. Thromb. Vasc. Biol. 2006, 26, 2056–2062. [Google Scholar] [CrossRef]
- Ali, M.T.; Martin, K.; Kumar, A.H.S.; Cavallin, E.; Pierrou, S.; Gleeson, B.M.; McPheat, W.L.; Turner, E.C.; Huang, C.-L.; Khider, W.; et al. A novel CX3CR1 antagonist eluting stent reduces stenosis by targeting inflammation. Biomaterials 2015, 69, 22–29. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Processes | References |
|---|---|
| Inflammation | [80,81,87,88,89] |
| Arteriolar remodeling and fibrosis | [90,91,92] |
| Capillary rarefaction or obliteration | [93,94,95] |
| Microthrombi (platelet activation) and hemostatic factors | [96,97,98] |
| Low shear stress/Atherogensis/Atherosclerosis | [77,80,99,100,101,102,103,104,105,106,107,108] |
| Endothelial/Vascular dysfunction | [90,91,92,93,94,109,110,111,112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stangret, A.; Sadowski, K.A.; Jabłoński, K.; Kochman, J.; Opolski, G.; Grabowski, M.; Tomaniak, M. Chemokine Fractalkine and Non-Obstructive Coronary Artery Disease—Is There a Link? Int. J. Mol. Sci. 2024, 25, 3885. https://doi.org/10.3390/ijms25073885
Stangret A, Sadowski KA, Jabłoński K, Kochman J, Opolski G, Grabowski M, Tomaniak M. Chemokine Fractalkine and Non-Obstructive Coronary Artery Disease—Is There a Link? International Journal of Molecular Sciences. 2024; 25(7):3885. https://doi.org/10.3390/ijms25073885
Chicago/Turabian StyleStangret, Aleksandra, Karol Artur Sadowski, Konrad Jabłoński, Janusz Kochman, Grzegorz Opolski, Marcin Grabowski, and Mariusz Tomaniak. 2024. "Chemokine Fractalkine and Non-Obstructive Coronary Artery Disease—Is There a Link?" International Journal of Molecular Sciences 25, no. 7: 3885. https://doi.org/10.3390/ijms25073885
APA StyleStangret, A., Sadowski, K. A., Jabłoński, K., Kochman, J., Opolski, G., Grabowski, M., & Tomaniak, M. (2024). Chemokine Fractalkine and Non-Obstructive Coronary Artery Disease—Is There a Link? International Journal of Molecular Sciences, 25(7), 3885. https://doi.org/10.3390/ijms25073885