
Machine Learning-Based Characterization and Identification of Tertiary Lymphoid Structures Using Spatial Transcriptomics Data


Abstract
:1. Introduction
2. Results
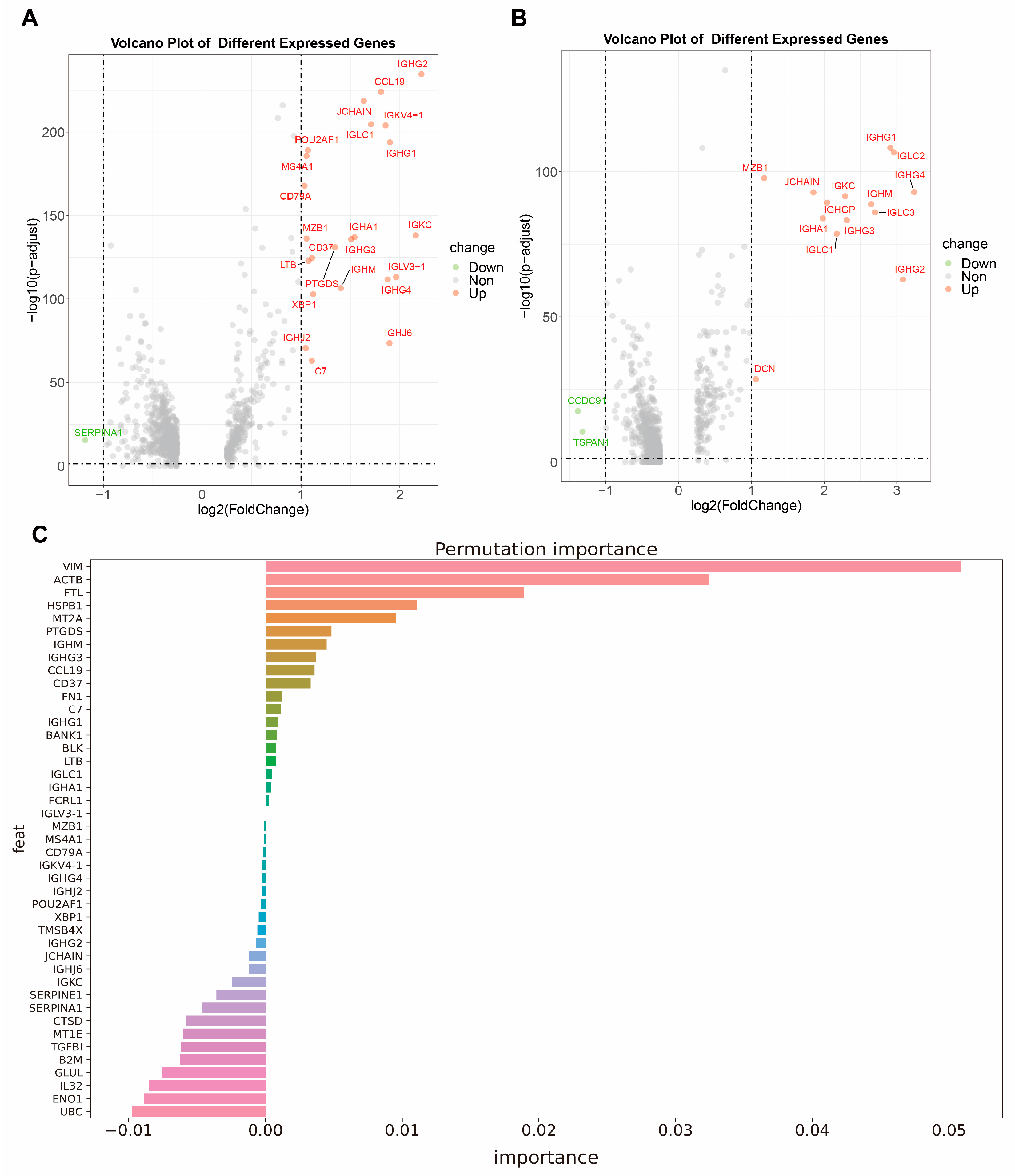
2.1. Gene Signatures Identified by Differential Expression
2.2. Gene Signatures Identified by Chi-Square Test
2.3. Markers of TLS Determined by Permutation Feature Importance
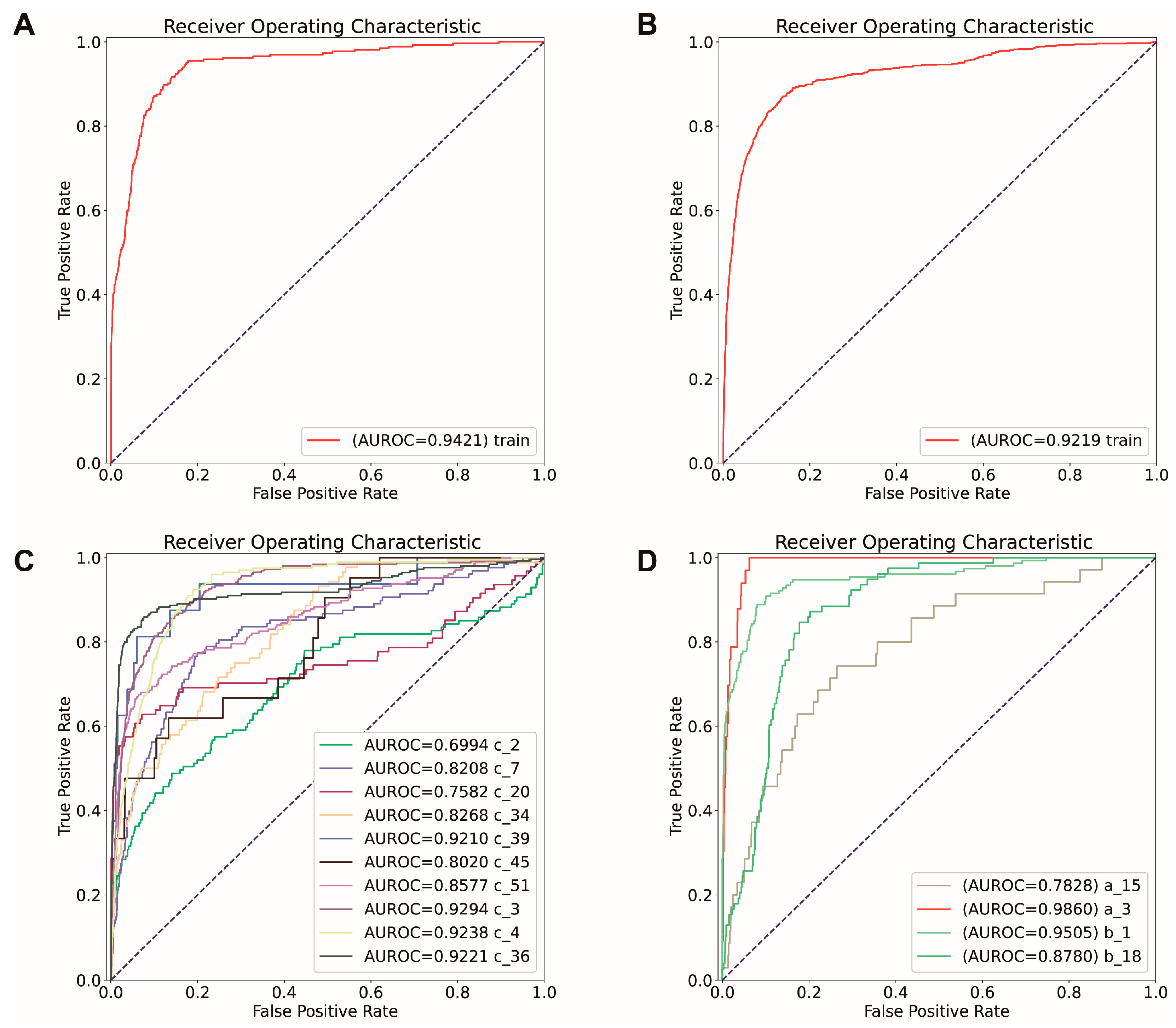
2.4. Construct SVC Models for TLS Prediction
2.5. Verify Markers’ Effectiveness by Model Comparison
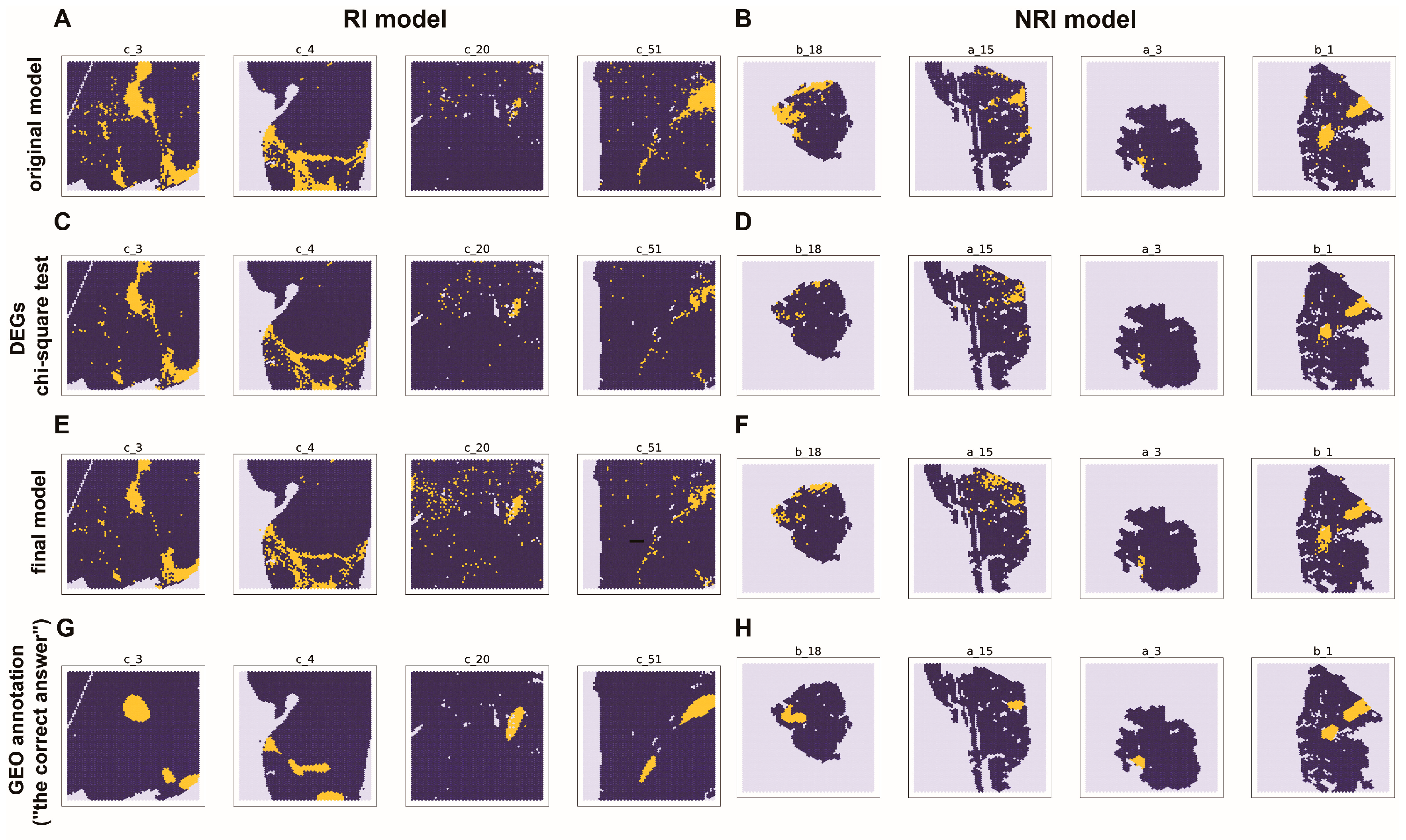
2.6. The Marker Genes and Their Spatial Distribution
3. Discussion
4. Materials and Methods
4.1. Data Source and Preprocessing
4.2. Data Selection for Model Construction
4.3. Model Construction and Performance Evaluation
4.4. Gene Signatures Identified by Differential Expression
4.5. Gene Signatures Selected by the Chi-Square Test
4.6. Gene Signatures Selected by Permutation Feature Importance
4.7. Spatial Distribution of Gene Signatures
4.8. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.C.; Goc, J.; Giraldo, N.A.; Sautès-Fridman, C.; Fridman, W.H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Trüb, M.; Zippelius, A. Tertiary lymphoid structures as a predictive biomarker of response to cancer immunotherapies. Front. Immunol. 2021, 12, 1432. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.S.; Nelson, B.H. Tumor-infiltrating B cells and T cells: Working together to promote patient survival. Oncoimmunology 2012, 1, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Goc, J.; Germain, C.; Vo-Bourgais, T.K.D.; Lupo, A.; Klein, C.; Knockaert, S.; de Chaisemartin, L.; Ouakrim, H.; Becht, E.; Alifano, M.; et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T CellsMature DC Coordinate Intratumoral Immune Reaction. Cancer Res. 2014, 74, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Di Caro, G.; Bergomas, F.; Grizzi, F.; Doni, A.; Bianchi, P.; Malesci, A.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. 2014, 20, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Posch, F.; Silina, K.; Leibl, S.; Mündlein, A.; Moch, H.; Siebenhüner, A.; Samaras, P.; Riedl, J.; Stotz, M.; Szkandera, J.; et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology 2018, 7, e1378844. [Google Scholar] [CrossRef]
- Hiraoka, N.; Ino, Y.; Yamazaki-Itoh, R.; Kanai, Y.; Kosuge, T.; Shimada, K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br. J. Cancer 2015, 112, 1782–1790. [Google Scholar] [CrossRef]
- Wirsing, A.M.; Ervik, I.K.; Seppola, M.; Uhlin-Hansen, L.; Steigen, S.E.; Hadler-Olsen, E. Presence of high-endothelial venules correlates with a favorable immune microenvironment in oral squamous cell carcinoma. Mod. Pathol. 2018, 31, 910–922. [Google Scholar] [CrossRef]
- Coppola, D.; Nebozhyn, M.; Khalil, F.; Dai, H.; Yeatman, T.; Loboda, A.; Mulé, J.J. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am. J. Pathol. 2011, 179, 37–45. [Google Scholar] [CrossRef]
- Gu-Trantien, C.; Loi, S.; Garaud, S.; Equeter, C.; Libin, M.; de Wind, A.; Ravoet, M.; Le Buanec, H.; Sibille, C.; Manfouo-Foutsop, G.; et al. CD4+follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Investig. 2013, 123, 2873–2892. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lee, S.J.; Ahn, J.; Park, W.Y.; Shin, D.H.; Lee, C.H.; Kwon, H.; Jeong, Y.J.; Ahn, H.Y.; I, H.; et al. The prognostic significance of tumor-infiltrating lymphocytes assessment with hematoxylin and eosin sections in resected primary lung adenocarcinoma. PLoS ONE 2019, 14, e0224430. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Marcoux, D.; Garaud, S.; Vitória, J.R.; Eynden, G.V.D.; de Wind, A.; De Silva, P.; Boisson, A.; Craciun, L.; Larsimont, D.; et al. BRCA gene mutations do not shape the extent and organization of tumor infiltrating lymphocytes in triple negative breast cancer. Cancer Lett. 2019, 450, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti–PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.L.; Fenstermacher, D.A.; Eschrich, S.; Qu, X.; Berglund, A.E.; Lloyd, M.C.; Schell, M.J.; Sondak, V.K.; Weber, J.S.; Mulé, J.J. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy? Sci. Rep. 2012, 2, 765. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, S.; Rizk, V.T.; Ma, Z.; Cheng, C.-H.; Berglund, A.E.; Coppola, D.; Khalil, F.; Mulé, J.J.; Soliman, H.H. Evaluation of invasive breast cancer samples using a 12-chemokine gene expression score: Correlation with clinical outcomes. Breast Cancer Res. 2017, 19, 71. [Google Scholar] [CrossRef]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy Distinct Immune Phenotypes of Colorectal Cancer Molecular Subtypes. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougoüin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541.e5. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Navai, N.; Alhalabi, O.; Siefker-Radtke, A.; Campbell, M.T.; Tidwell, R.S.; Guo, C.C.; Kamat, A.M.; Matin, S.F.; Araujo, J.C.; et al. Neoadjuvant PD-L1 plus CTLA-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nat. Med. 2020, 26, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef]
- Johansson-Percival, A.; He, B.; Li, Z.J.; Kjellén, A.; Russell, K.; Li, J.; Larma, I.; Ganss, R. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol. 2017, 18, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.-M.; Italiano, A.; Sautès-Fridman, C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, C.S.; Fontugne, J.; Cabel, L.; Bernard-Pierrot, I.; Radvanyi, F.; Allory, Y.; de Reyniès, A. Tertiary lymphoid structures marker CXCL13 is associated with better survival for patients with advanced-stage bladder cancer treated with immunotherapy. Eur. J. Cancer 2021, 148, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, A.; Derangere, V.; Boidot, R.; Apetoh, L.; Vincent, J.; Orry, D.; Fraisse, J.; Causeret, S.; Martin, F.; Arnould, L.; et al. Tumor infiltration by Tbet+ effector T cells and CD20+ B cells is associated with survival in gastric cancer patients. Oncoimmunology 2016, 5, e1054598. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Garrido, I.; Filleron, T.; Le Guellec, S.; Bellard, E.; Fournie, J.-J.; Rochaix, P.; Girard, J.-P. Human solid tumors contain high endothelial venules: Association with T-and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res. 2011, 71, 5678–5687. [Google Scholar] [CrossRef]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer Plasma Cells, CD8 T Cells, and Survival in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef]
- Bao, Q.; Guo, X.; Cao, C.; Li, Q.-Y.; Sun, L.; Ye, X.-Y.; Li, L.-Y.; Dong, J.-C.; Gao, Y.-F.; Chen, H.-X.; et al. Presence of tertiary lymphoid organ in nasal inverted papilloma is correlated with eosinophil infiltration and local immunoglobulin production. Int. Arch. Allergy Immunol. 2021, 182, 350–359. [Google Scholar] [CrossRef]
- Liu, Z. A method of SVM with normalization in intrusion detection. Procedia Environ. Sci. 2011, 11, 256–262. [Google Scholar] [CrossRef]
- Perez, M.; Rubin, D.M.; Marwala, T.; Scott, L.E.; Featherston, J.; Stevens, W. The fuzzy gene filter: An adaptive fuzzy inference system for expression array feature selection. In Proceedings of the Trends in Applied Intelligent Systems: 23rd International Conference on Industrial Engineering and Other Applications of Applied Intelligent Systems, IEA/AIE 2010, Cordoba, Spain, 1–4 June 2010; Proceedings, Part III 23. Springer: Berlin/Heidelberg, Germany, 2010; pp. 62–71. [Google Scholar] [CrossRef]
- Cherrington, M.; Thabtah, F.; Lu, J.; Xu, Q. Feature selection: Filter methods performance challenges. In Proceedings of the 2019 International Conference on Computer and Information Sciences (ICCIS), Sakaka, Saudi Arabia, 3–4 April 2019; pp. 1–4. [Google Scholar] [CrossRef]
- Altmann, A.; Toloşi, L.; Sander, O.; Lengauer, T. Permutation importance: A corrected feature importance measure. Bioinformatics 2010, 26, 1340–1347. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method/Model | RI | NRI |
|---|---|---|
| Differential expression | IGHG2, CCL19, JCHAIN, IGLC1, IGKV4-1, IGHG1, POU2AF1, MS4A1, CD79A, IGKC, IGHA1, MZB1, IGHG3, PTGDS, CD37, LTB, IGLV3-1, IGHG4, IGHM, XBP1, IGHJ6, IGHJ2, C7, SERPINA1 | IGHG1, IGLC2, MZB1, IGHG4, JCHAIN, IGKC, IGHGP, IGHM, IGLC3, IGHA1, IGHG3, IGLC1, IGLV4-69, IGHG2, DCN, CCDC91, TSPAN1 |
| Chi-square test | IL32, HSPB1, B2M, JCHAIN, TGFBI, MS4A1, GLUL, BLK, IGKV4-1, VIM, IGHG1, FN1, IGHG2, C7, CCL19, MT2A, FTL, BANK1, IGKC, MT1E, IGLV3-1, FCRL1, ACTB, UBC, IGLC1, IGHG3, TMSB4X, ENO1, CTSD, SERPINE1 | RPL37, HLA-B, SPP1, CD74, IGLC3, TMSB10, VIM, RPL37A, RPL34, RPL41, NDRG1, HLA-A, IGLC2, RPL13, RPS8, IGFBP7, RPL10, RPLP1, TGFBI, B2M, RPS18, RPS27, TPT1, FTH1, MIF, IGKC, RPS2, FTL, RPL39, EEF1A1, CD24, ITM2B, RPS23, GAPDH, IGHG2, RPS21, RPL36, IGHG1 |
| Permutation importance | HSPB1, LTB, FTL, VIM, BLK, IGLC1, C7, IGHG1, FCRL1, PTGDS, IGHG3, IGHA1, FN1, IGLV3-1, ACTB, BANK1, MT2A, CCL19, IGHM, CD37 | TGFBI, TPT1, FTL, IGHG4, IGKC, IGLC1, EEF1A1, IGLC3, IGHGP, IGLC2, DCN, IGHG2, RPS27, VIM, IGHG3, FTH1, IGHA1 |
| Final markers | HSPB1, LTB, FTL, VIM, BLK, IGLC1, C7, IGHG1, FCRL1, PTGDS, IGHG3, IGHA1, FN1, IGLV3-1, ACTB, BANK1, MT2A, CCL19, IGHM, CD37 | TGFBI, TPT1, FTL, IGHG4, IGKC, IGLC1, EEF1A1, IGLC3, IGHGP, IGLC2, DCN, IGHG2, RPS27, VIM, IGHG3, FTH1, IGHA1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Wang, Z.; Huang, H.-D.; Lee, T.-Y. Machine Learning-Based Characterization and Identification of Tertiary Lymphoid Structures Using Spatial Transcriptomics Data. Int. J. Mol. Sci. 2024, 25, 3887. https://doi.org/10.3390/ijms25073887
Li S, Wang Z, Huang H-D, Lee T-Y. Machine Learning-Based Characterization and Identification of Tertiary Lymphoid Structures Using Spatial Transcriptomics Data. International Journal of Molecular Sciences. 2024; 25(7):3887. https://doi.org/10.3390/ijms25073887
Chicago/Turabian StyleLi, Songyun, Zhuo Wang, Hsien-Da Huang, and Tzong-Yi Lee. 2024. "Machine Learning-Based Characterization and Identification of Tertiary Lymphoid Structures Using Spatial Transcriptomics Data" International Journal of Molecular Sciences 25, no. 7: 3887. https://doi.org/10.3390/ijms25073887
APA StyleLi, S., Wang, Z., Huang, H. -D., & Lee, T. -Y. (2024). Machine Learning-Based Characterization and Identification of Tertiary Lymphoid Structures Using Spatial Transcriptomics Data. International Journal of Molecular Sciences, 25(7), 3887. https://doi.org/10.3390/ijms25073887