
Oleate Promotes Triple-Negative Breast Cancer Cell Migration by Enhancing Filopodia Formation through a PLD/Cdc42-Dependent Pathway



Abstract
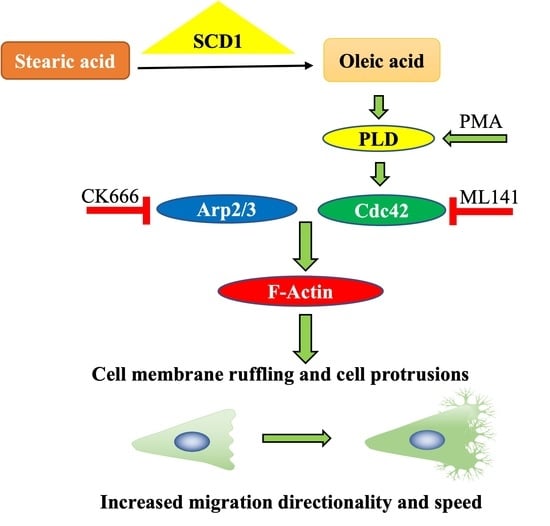
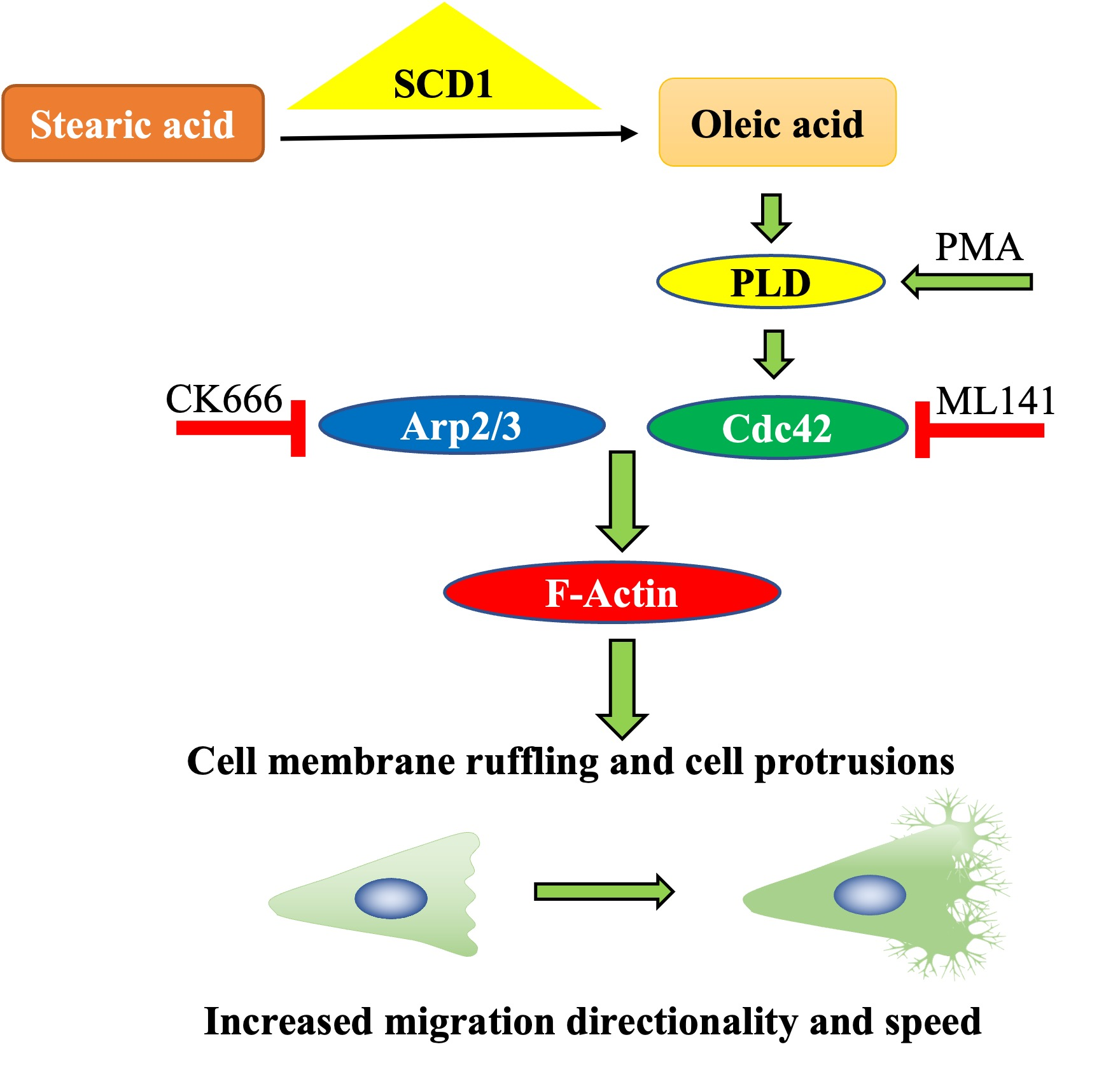
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
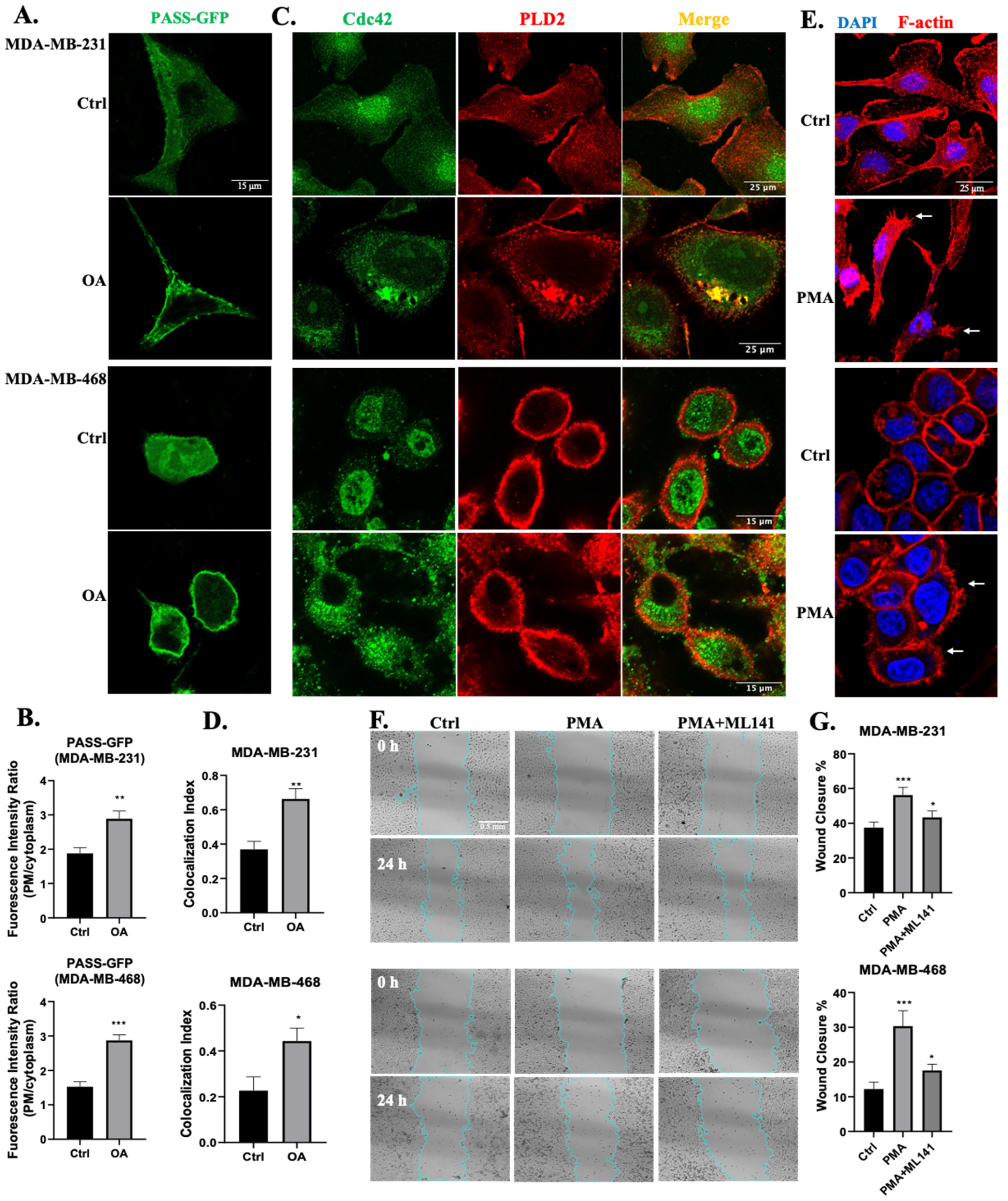
2.1. OA-Induced Cell Membrane Ruffling in TNBC Cells
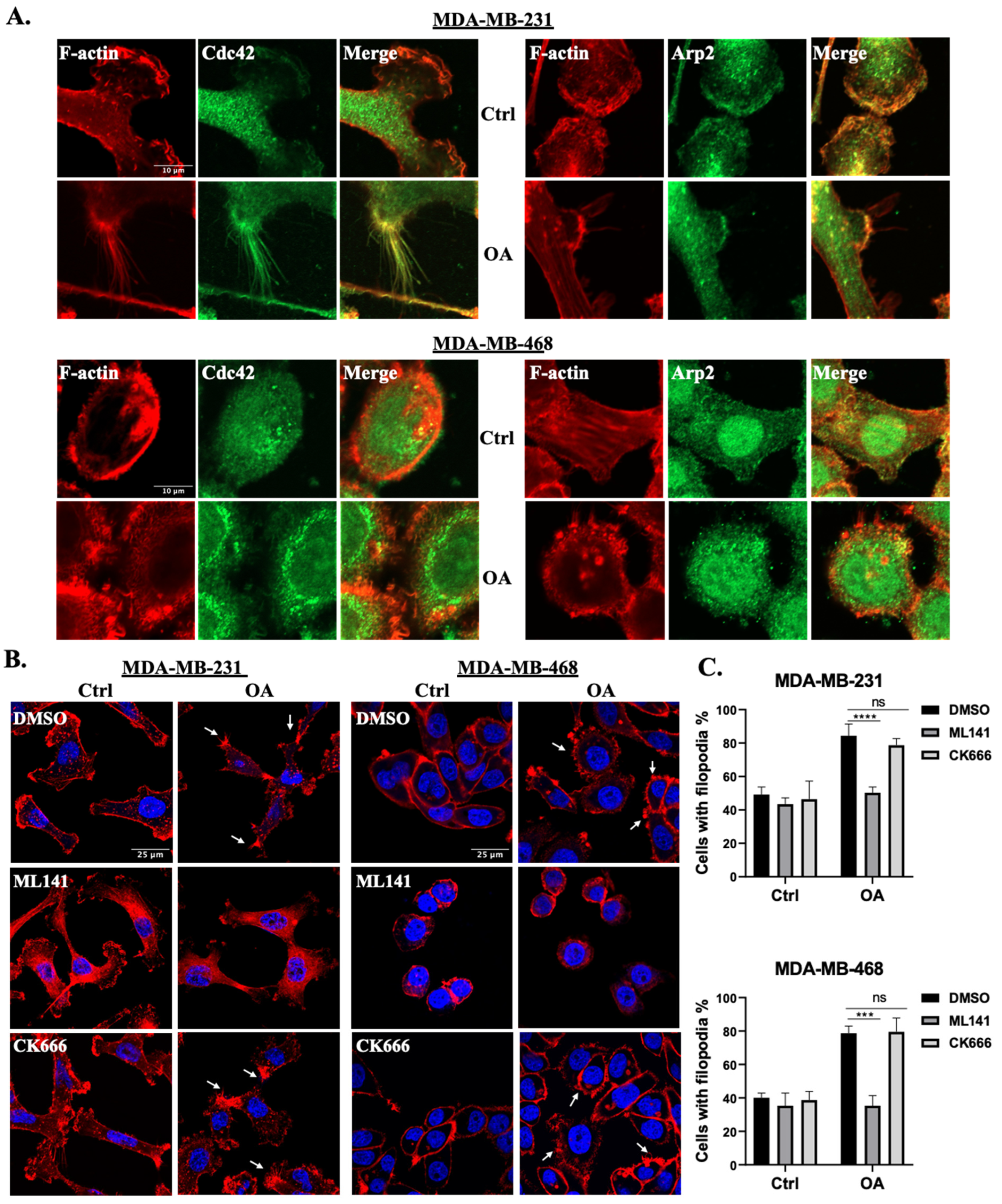
2.2. OA-Induced Translocation of Cdc42 and the Arp2/3 Complex in TNBC Cells
2.3. Cdc42 Activity Is Required for OA-Induced Filopodia Formation in TNBC Cells
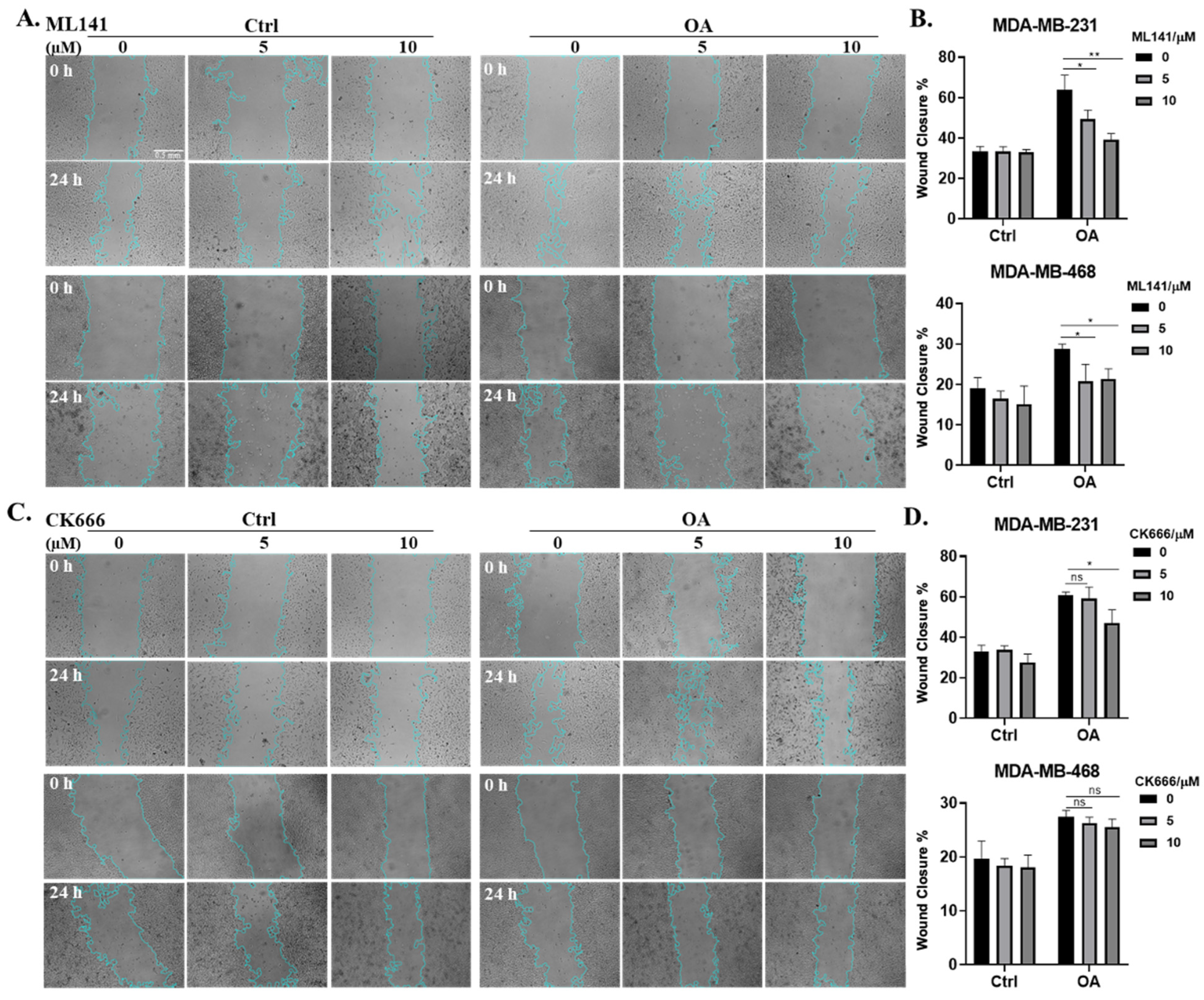
2.4. Cdc42 Activity Is Required for OA-Induced Cell Migration in TNBC Cells
2.5. PLD Is Involved in OA-Induced Filopodia Formation and Cell Migration
2.6. Elevated CDC42 Expression and Its Association with Survival in TNBC Patients
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Transfection
4.3. Immunofluorescence Staining and F-Actin Staining
4.4. Confocal Microscopy and Image Analysis
4.5. Wound Healing Assay
4.6. Cell Viability Assay
4.7. Bioinformatic Analyses
4.8. Western Blotting
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giaquinto:, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Eliyatkin, N.; Yalcin, E.; Zengel, B.; Aktas, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to a New Age, and a New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Devericks, E.N.; Carson, M.S.; McCullough, L.E.; Coleman, M.F.; Hursting, S.D. The obesity-breast cancer link: A multidisciplinary perspective. Cancer Metastasis Rev. 2022, 41, 607–625. [Google Scholar] [CrossRef]
- Landry, I.; Sumbly, V.; Vest, M. Advancements in the Treatment of Triple-Negative Breast Cancer: A Narrative Review of the Literature. Cureus 2022, 14, e21970. [Google Scholar] [CrossRef]
- Prendeville, H.; Lynch, L. Diet, lipids, and antitumor immunity. Cell. Mol. Immunol. 2022, 19, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Gunstone, F.D. Fatty acids—Nomenclature, structure, isolation and structure determination, biosynthesis and chemical synthesis. In Fatty Acid and Lipid Chemistry; Gunstone, F.D., Ed.; Springer: Boston, MA, USA, 1996; pp. 1–34. [Google Scholar]
- Davis, C.; Bryan, J.; Hodgson, J.; Murphy, K. Definition of the Mediterranean Diet; A Literature Review. Nutrients 2015, 7, 9139–9153. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Schwedhelm, C.; Galbete, C.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: An Updated Systematic Review and Meta-Analysis. Nutrients 2017, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Morze, J.; Danielewicz, A.; Przybylowicz, K.; Zeng, H.; Hoffmann, G.; Schwingshackl, L. An updated systematic review and meta-analysis on adherence to mediterranean diet and risk of cancer. Eur. J. Nutr. 2021, 60, 1561–1586. [Google Scholar] [CrossRef]
- Assy, N.; Nassar, F.; Nasser, G.; Grosovski, M. Olive oil consumption and non-alcoholic fatty liver disease. World J. Gastroenterol. 2009, 15, 1809–1815. [Google Scholar] [CrossRef]
- Psaltopoulou, T.; Kosti, R.I.; Haidopoulos, D.; Dimopoulos, M.; Panagiotakos, D.B. Olive oil intake is inversely related to cancer prevalence: A systematic review and a meta-analysis of 13,800 patients and 23,340 controls in 19 observational studies. Lipids Health Dis. 2011, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Bergeron, K.F.; Lingrand, M.; Mounier, C. Unveiling the MUFA-Cancer Connection: Insights from Endogenous and Exogenous Perspectives. Int. J. Mol. Sci. 2023, 24, 9921. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, J.D.; Subramanian, T.; Liu, K.; King, M.R. Rafting Down the Metastatic Cascade: The Role of Lipid Rafts in Cancer Metastasis, Cell Death, and Clinical Outcomes. Cancer Res. 2021, 81, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guan, L.; Li, S.; Jiang, Y.; Xiong, N.; Li, L.; Wu, C.; Zeng, H.; Liu, Y. Mechanosensitive caveolin-1 activation-induced PI3K/Akt/mTOR signaling pathway promotes breast cancer motility, invadopodia formation and metastasis in vivo. Oncotarget 2016, 7, 16227–16247. [Google Scholar] [CrossRef]
- Wu, J.S.; Jiang, J.; Chen, B.J.; Wang, K.; Tang, Y.L.; Liang, X.H. Plasticity of cancer cell invasion: Patterns and mechanisms. Transl. Oncol. 2021, 14, 100899. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Zech, T. Actin-Based Cell Protrusion in a 3D Matrix. Trends Cell Biol. 2018, 28, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Bonfim-Melo, A.; Ferreira, E.R.; Mortara, R.A. Rac1/WAVE2 and Cdc42/N-WASP Participation in Actin-Dependent Host Cell Invasion by Extracellular Amastigotes of Trypanosoma cruzi. Front. Microbiol. 2018, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Randzavola, L.O.; Strege, K.; Juzans, M.; Asano, Y.; Stinchcombe, J.C.; Gawden-Bone, C.M.; Seaman, M.N.; Kuijpers, T.W.; Griffiths, G.M. Loss of ARPC1B impairs cytotoxic T lymphocyte maintenance and cytolytic activity. J. Clin. Investig. 2019, 129, 5600–5614. [Google Scholar] [CrossRef]
- Le Clainche, C.; Carlier, M.F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 2008, 88, 489–513. [Google Scholar] [CrossRef]
- He, S.; Ji, B. Mechanics of Cell Mechanosensing in Protrusion and Retraction of Lamellipodium. ACS Biomater. Sci. Eng. 2017, 3, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, M. New insights into the formation and the function of lamellipodia and ruffles in mesenchymal cell migration. Cell Adhes. Migr. 2018, 12, 401–416. [Google Scholar] [CrossRef] [PubMed]
- San Miguel-Ruiz, J.E.; Letourneau, P.C. The role of Arp2/3 in growth cone actin dynamics and guidance is substrate dependent. J. Neurosci. 2014, 34, 5895–5908. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Ellis, A.; Sepulveda-Ramirez, S.P.; Salgado, T.; Terrazas, I.; Reyes, G.; De La Rosa, R.; Henson, J.H.; Shuster, C.B. Rac and Arp2/3-Nucleated Actin Networks Antagonize Rho During Mitotic and Meiotic Cleavages. Front. Cell Dev. Biol. 2020, 8, 591141. [Google Scholar] [CrossRef] [PubMed]
- Shakir, M.A.; Jiang, K.; Struckhoff, E.C.; Demarco, R.S.; Patel, F.B.; Soto, M.C.; Lundquist, E.A. The Arp2/3 activators WAVE and WASP have distinct genetic interactions with Rac GTPases in Caenorhabditis elegans axon guidance. Genetics 2008, 179, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef]
- Nolen, B.J.; Littlefield, R.S.; Pollard, T.D. Crystal structures of actin-related protein 2/3 complex with bound ATP or ADP. Proc. Natl. Acad. Sci. USA 2004, 101, 15627–15632. [Google Scholar] [CrossRef]
- Hoon, J.L.; Wong, W.K.; Koh, C.G. Functions and regulation of circular dorsal ruffles. Mol. Cell. Biol. 2012, 32, 4246–4257. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, M.C.; Bogdan, S. Collective cell migration driven by filopodia-New insights from the social behavior of myotubes. Bioessays 2021, 43, e2100124. [Google Scholar] [CrossRef]
- Gat, S.; Simon, C.; Campillo, C.; Bernheim-Groswasser, A.; Sykes, C. Finger-like membrane protrusions are favored by heterogeneities in the actin network. Soft Matter 2020, 16, 7222–7230. [Google Scholar] [CrossRef]
- Bray, K.; Gillette, M.; Young, J.; Loughran, E.; Hwang, M.; Sears, J.C.; Vargo-Gogola, T. Cdc42 overexpression induces hyperbranching in the developing mammary gland by enhancing cell migration. Breast Cancer Res. 2013, 15, R91. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, M.; Fan, J.; Odaka, Y.; Liu, N.; Hassan, A.; Duan, X.; Stump, P.; Byerly, L.; Donaldson, M.; Hao, J.; et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 2017, 114, 10918–10923. [Google Scholar] [CrossRef]
- Bischoff, M.C.; Lieb, S.; Renkawitz-Pohl, R.; Bogdan, S. Filopodia-based contact stimulation of cell migration drives tissue morphogenesis. Nat. Commun. 2021, 12, 791. [Google Scholar] [CrossRef] [PubMed]
- Kiso, M.; Tanaka, S.; Saji, S.; Toi, M.; Sato, F. Long isoform of VEGF stimulates cell migration of breast cancer by filopodia formation via NRP1/ARHGAP17/Cdc42 regulatory network. Int. J. Cancer 2018, 143, 2905–2918. [Google Scholar] [CrossRef] [PubMed]
- Arjonen, A.; Kaukonen, R.; Ivaska, J. Filopodia and adhesion in cancer cell motility. Cell Adhes. Migr. 2011, 5, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Tomishige, N.; Murate, M.; Ishitsuka, R.; Schaller, H.; Mely, Y.; Ueda, K.; Kobayashi, T. Cholesterol asymmetry at the tip of filopodia during cell adhesion. FASEB J. 2020, 34, 6185–6197. [Google Scholar] [CrossRef]
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr. Opin. Cell Biol. 2015, 36, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Merino-Casallo, F.; Gomez-Benito, M.J.; Hervas-Raluy, S.; Garcia-Aznar, J.M. Unravelling cell migration: Defining movement from the cell surface. Cell Adhes. Migr. 2022, 16, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Kang, D.; Ji, D.; Wang, X.; Zhan, W.; Fu, M.; Xin, H.B.; Wang, J.B. How does cancer cell metabolism affect tumor migration and invasion? Cell Adhes. Migr. 2013, 7, 395–403. [Google Scholar] [CrossRef]
- Marcial-Medina, C.; Ordonez-Moreno, A.; Gonzalez-Reyes, C.; Cortes-Reynosa, P.; Perez Salazar, E. Oleic acid induces migration through a FFAR1/4, EGFR and AKT-dependent pathway in breast cancer cells. Endocr. Connect. 2019, 8, 252–265. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, Y.; Yuan, Y.; Zhang, X.; Qin, C.; Xie, J.; Hao, Y.; Xu, T.; Wang, X. Effects of oleic acid on cell proliferation through an integrin-linked kinase signaling pathway in 786-O renal cell carcinoma cells. Oncol. Lett. 2013, 5, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Qing, C.P.; Sheng, Z.Z.; Li, G.Z.; Man, L.B.; Xu, T. Oleic acid promotes cell invasion through an integrin-linked kinase signaling pathway in renal cell carcinoma. Nutr. Cancer 2020, 72, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Soto-Guzman, A.; Navarro-Tito, N.; Castro-Sanchez, L.; Martinez-Orozco, R.; Salazar, E.P. Oleic acid promotes MMP-9 secretion and invasion in breast cancer cells. Clin. Exp. Metastasis 2010, 27, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tito, N.; Soto-Guzman, A.; Castro-Sanchez, L.; Martinez-Orozco, R.; Salazar, E.P. Oleic acid promotes migration on MDA-MB-231 breast cancer cells through an arachidonic acid-dependent pathway. Int. J. Biochem. Cell Biol. 2010, 42, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Soto-Guzman, A.; Villegas-Comonfort, S.; Cortes-Reynosa, P.; Perez Salazar, E. Role of arachidonic acid metabolism in Stat5 activation induced by oleic acid in MDA-MB-231 breast cancer cells. Prostaglandins Leukot Essent Fat. Acids 2013, 88, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.J.; Chan, S.H.; Lee, C.T.; Huang, W.C.; Tsai, J.P.; Chen, B.K. Oleic acid-induced ANGPTL4 enhances head and neck squamous cell carcinoma anoikis resistance and metastasis via up-regulation of fibronectin. Cancer Lett. 2017, 386, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Lingrand, M.; Lalonde, S.; Jutras-Carignan, A.; Bergeron, K.F.; Rassart, E.; Mounier, C. SCD1 activity promotes cell migration via a PLD-mTOR pathway in the MDA-MB-231 triple-negative breast cancer cell line. Breast Cancer 2020, 27, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.P.; Binti Ahmad Mokhtar, A.M.; Mott, H.R.; Owen, D. Molecular subversion of Cdc42 signalling in cancer. Biochem. Soc. Trans. 2021, 49, 1425–1442. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Tay, L.W.; He, J.; Du, G. Monitoring Phosphatidic Acid Signaling in Breast Cancer Cells Using Genetically Encoded Biosensors. Methods Mol. Biol. 2016, 1406, 225–237. [Google Scholar] [CrossRef]
- Bruntz, R.C.; Lindsley, C.W.; Brown, H.A. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol. Rev. 2014, 66, 1033–1079. [Google Scholar] [CrossRef]
- Vinggaard, A.M.; Hansen, H.S. Phorbol ester and vasopressin activate phospholipase D in Leydig cells. Mol. Cell. Endocrinol. 1991, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, Z.; Zhang, W.; Ye, Z.; Liu, F. GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 2021, 49, W242–W246. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Jacquemet, G.; Paatero, I.; Carisey, A.F.; Padzik, A.; Orange, J.S.; Hamidi, H.; Ivaska, J. FiloQuant reveals increased filopodia density during breast cancer progression. J. Cell Biol. 2017, 216, 3387–3403. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Zhou, X.; Li, L.; Liu, Q.; Wang, Z.; Bai, X.; Zhao, Y.; Shi, H.; Zhang, X.; et al. The oncoprotein HBXIP enhances migration of breast cancer cells through increasing filopodia formation involving MEKK2/ERK1/2/Capn4 signaling. Cancer Lett. 2014, 355, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perala, M.; Kronqvist, P.; Joensuu, H.; et al. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun. 2016, 7, 13297. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, H.; Zhao, W.; Meng, Y.; Li, J.; Huang, T.; Sun, J. Cdc42-mediated supracellular cytoskeleton induced cancer cell migration under low shear stress. Biochem. Biophys. Res. Commun. 2019, 519, 134–140. [Google Scholar] [CrossRef]
- Aikemu, B.; Shao, Y.; Yang, G.; Ma, J.; Zhang, S.; Yang, X.; Hong, H.; Yesseyeva, G.; Huang, L.; Jia, H.; et al. NDRG1 regulates Filopodia-induced Colorectal Cancer invasiveness via modulating CDC42 activity. Int. J. Biol. Sci. 2021, 17, 1716–1730. [Google Scholar] [CrossRef] [PubMed]
- Horita, K.; Kurosaki, H.; Nakatake, M.; Kuwano, N.; Oishi, T.; Itamochi, H.; Sato, S.; Kono, H.; Ito, M.; Hasegawa, K.; et al. lncRNA UCA1-Mediated Cdc42 Signaling Promotes Oncolytic Vaccinia Virus Cell-to-Cell Spread in Ovarian Cancer. Mol. Ther. Oncolytics 2019, 13, 35–48. [Google Scholar] [CrossRef]
- Horita, K.; Kurosaki, H.; Nakatake, M.; Ito, M.; Kono, H.; Nakamura, T. Long noncoding RNA UCA1 enhances sensitivity to oncolytic vaccinia virus by sponging miR-18a/miR-182 and modulating the Cdc42/filopodia axis in colorectal cancer. Biochem. Biophys. Res. Commun. 2019, 516, 831–838. [Google Scholar] [CrossRef]
- Yuan, Z.; Wei, W. RAB5A promotes the formation of filopodia in pancreatic cancer cells via the activation of cdc42 and beta1-integrin. Biochem. Biophys. Res. Commun. 2021, 535, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.H.; Chao, Y.C.; Hung, P.F.; Chen, H.Y.; Yang, S.C.; Chang, Y.L.; Wu, C.T.; Chang, C.C.; Wang, W.L.; Chan, W.K.; et al. The ability of LCRMP-1 to promote cancer invasion by enhancing filopodia formation is antagonized by CRMP-1. J. Clin. Investig. 2011, 121, 3189–3205. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Medina, J.I.; Velazquez, L.; Dharmawardhane, S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front. Cell Dev. Biol. 2020, 8, 201. [Google Scholar] [CrossRef] [PubMed]
- Dubash, A.D.; Guilluy, C.; Srougi, M.C.; Boulter, E.; Burridge, K.; Garcia-Mata, R. The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS ONE 2011, 6, e17380. [Google Scholar] [CrossRef] [PubMed]
- Liotti, A.; Cosimato, V.; Mirra, P.; Cali, G.; Conza, D.; Secondo, A.; Luongo, G.; Terracciano, D.; Formisano, P.; Beguinot, F.; et al. Oleic acid promotes prostate cancer malignant phenotype via the G protein-coupled receptor FFA1/GPR40. J. Cell. Physiol. 2018, 233, 7367–7378. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Nagaya, H.; Kanno, T.; Nishizaki, T. Oleic acid stimulates glucose uptake into adipocytes by enhancing insulin receptor signaling. J. Pharmacol. Sci. 2014, 126, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Wirth, A.; Ponimaskin, E. Lipidation of small GTPase Cdc42 as regulator of its physiological and pathophysiological functions. Front. Physiol. 2022, 13, 1088840. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Linder, M.E. Identification of a novel prenyl and palmitoyl modification at the CaaX motif of Cdc42 that regulates RhoGDI binding. Mol. Cell. Biol. 2013, 33, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.; Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J Cancer 2002, 87, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.C.; Zhang, Y.; Qu, X.C. Effects of Cdc42 overexpression on the estrogen-enhanced multidrug resistance in breast cancer cells. Zhonghua Zhong Liu Za Zhi 2011, 33, 489–493. [Google Scholar] [PubMed]
- Du, D.S.; Yang, X.Z.; Wang, Q.; Dai, W.J.; Kuai, W.X.; Liu, Y.L.; Chu, D.; Tang, X.J. Effects of CDC42 on the proliferation and invasion of gastric cancer cells. Mol. Med. Rep. 2016, 13, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Feng, J.G.; Tuersun, A.; Liu, T.; Liu, H.; Liu, Q.; Zheng, S.T.; Huang, C.G.; Lv, G.D.; Sheyhidin, I.; et al. Proteomic identification of differentially-expressed proteins in esophageal cancer in three ethnic groups in Xinjiang. Mol. Biol. Rep. 2011, 38, 3261–3269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Zhang, Y.; Miao, Y.; Zhao, Y.; Zhang, P.X.; Jiang, G.Y.; Zhang, J.Y.; Han, Y.; Lin, X.Y.; et al. Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung Cancer 2009, 63, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Jiao, D.M.; Yao, Q.H.; Yan, J.; Song, J.; Chen, F.Y.; Lu, G.H.; Zhou, J.Y. Expression analysis of Cdc42 in lung cancer and modulation of its expression by curcumin in lung cancer cell lines. Int. J. Oncol. 2012, 40, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, H.; Liu, N.; Wang, L.; Wang, E. Correlation of expression of p120ctn, RhoA and Cdc42 and their significance in non-small cell lung cancer. Zhongguo Fei Ai Za Zhi 2005, 8, 304–308. [Google Scholar] [CrossRef]
- Tucci, M.G.; Lucarini, G.; Brancorsini, D.; Zizzi, A.; Pugnaloni, A.; Giacchetti, A.; Ricotti, G.; Biagini, G. Involvement of E-cadherin, beta-catenin, Cdc42 and CXCR4 in the progression and prognosis of cutaneous melanoma. Br. J. Dermatol. 2007, 157, 1212–1216. [Google Scholar] [CrossRef]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Gilad, E.; Rothman, K.; Peyrollier, K. Hyaluronan-CD44 interaction with IQGAP1 promotes Cdc42 and ERK signaling, leading to actin binding, Elk-1/estrogen receptor transcriptional activation, and ovarian cancer progression. J. Biol. Chem. 2005, 280, 11961–11972. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, Y.; Cheng, Y.; Hong, L.; Wang, C.; Wei, Z.; Cai, Q.; Yan, R. High Expression of Cell Division Cycle 42 Promotes Pancreatic Cancer Growth and Predicts Poor Outcome of Pancreatic Cancer Patients. Dig. Dis. Sci. 2017, 62, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Kamai, T.; Yamanishi, T.; Shirataki, H.; Takagi, K.; Asami, H.; Ito, Y.; Yoshida, K. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 2004, 10, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Thomas, P.G.; Lindsley, C.W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Bowling, F.Z.; Frohman, M.A.; Airola, M.V. Structure and regulation of human phospholipase D. Adv. Biol. Regul. 2021, 79, 100783. [Google Scholar] [CrossRef] [PubMed]
- Henkels, K.M.; Boivin, G.P.; Dudley, E.S.; Berberich, S.J.; Gomez-Cambronero, J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene 2013, 32, 5551–5562. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, Y.; Lee, S.D.; Lopez, I.; Arnold, R.S.; Lambeth, J.D.; Suh, P.G.; Ryu, S.H. Selective activation of phospholipase D2 by unsaturated fatty acid. FEBS Lett. 1999, 454, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Ohguchi, K.; Nakashima, S.; Ito, Y.; Naganawa, T.; Kondo, N.; Nozawa, Y. Increased activity of oleate-dependent type phospholipase D during actinomycin D-induced apoptosis in Jurkat T cells. J. Immunol. 1998, 161, 6469–6474. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, T.C.; Meier, K.E. Expression and regulation of phospholipase D isoforms in mammalian cell lines. J. Cell. Physiol. 2000, 182, 77–87. [Google Scholar] [CrossRef]
- Sarri, E.; Pardo, R.; Fensome-Green, A.; Cockcroft, S. Endogenous phospholipase D2 localizes to the plasma membrane of RBL-2H3 mast cells and can be distinguished from ADP ribosylation factor-stimulated phospholipase D1 activity by its specific sensitivity to oleic acid. Biochem. J. 2003, 369, 319–329. [Google Scholar] [CrossRef]
- O’Luanaigh, N.; Pardo, R.; Fensome, A.; Allen-Baume, V.; Jones, D.; Holt, M.R.; Cockcroft, S. Continual production of phosphatidic acid by phospholipase D is essential for antigen-stimulated membrane ruffling in cultured mast cells. Mol. Biol. Cell 2002, 13, 3730–3746. [Google Scholar] [CrossRef]
- Colley, W.C.; Sung, T.C.; Roll, R.; Jenco, J.; Hammond, S.M.; Altshuller, Y.; Bar-Sagi, D.; Morris, A.J.; Frohman, M.A. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 1997, 7, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zheng, Y.; Foster, D.A. Phospholipase D2 stimulates cell protrusion in v-Src-transformed cells. Biochem. Biophys. Res. Commun. 2002, 293, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Mahankali, M.; Peng, H.J.; Henkels, K.M.; Dinauer, M.C.; Gomez-Cambronero, J. Phospholipase D2 (PLD2) is a guanine nucleotide exchange factor (GEF) for the GTPase Rac2. Proc. Natl. Acad. Sci. USA 2011, 108, 19617–19622. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.J.; Henkels, K.M.; Mahankali, M.; Dinauer, M.C.; Gomez-Cambronero, J. Evidence for two CRIB domains in phospholipase D2 (PLD2) that the enzyme uses to specifically bind to the small GTPase Rac2. J. Biol. Chem. 2011, 286, 16308–16320. [Google Scholar] [CrossRef]
- Yang, C.; Svitkina, T. Filopodia initiation: Focus on the Arp2/3 complex and formins. Cell Adhes. Migr. 2011, 5, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Condon, N.D.; Stow, J.L.; Wall, A.A. Automated Analysis of Cell Surface Ruffling: Ruffle Quantification Macro. Bio Protoc. 2020, 10, e3494. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbelaez, P.; Cruz, J.C.; Munoz-Camargo, C. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Bergeron, K.-F.; Mounier, C. Oleate Promotes Triple-Negative Breast Cancer Cell Migration by Enhancing Filopodia Formation through a PLD/Cdc42-Dependent Pathway. Int. J. Mol. Sci. 2024, 25, 3956. https://doi.org/10.3390/ijms25073956
Guo Z, Bergeron K-F, Mounier C. Oleate Promotes Triple-Negative Breast Cancer Cell Migration by Enhancing Filopodia Formation through a PLD/Cdc42-Dependent Pathway. International Journal of Molecular Sciences. 2024; 25(7):3956. https://doi.org/10.3390/ijms25073956
Chicago/Turabian StyleGuo, Zhiqiang, Karl-Frédérik Bergeron, and Catherine Mounier. 2024. "Oleate Promotes Triple-Negative Breast Cancer Cell Migration by Enhancing Filopodia Formation through a PLD/Cdc42-Dependent Pathway" International Journal of Molecular Sciences 25, no. 7: 3956. https://doi.org/10.3390/ijms25073956
APA StyleGuo, Z., Bergeron, K. -F., & Mounier, C. (2024). Oleate Promotes Triple-Negative Breast Cancer Cell Migration by Enhancing Filopodia Formation through a PLD/Cdc42-Dependent Pathway. International Journal of Molecular Sciences, 25(7), 3956. https://doi.org/10.3390/ijms25073956