
Nuclear MAST4 Suppresses FOXO3 through Interaction with AKT3 and Induces Chemoresistance in Pancreatic Ductal Carcinoma
 ,
,
Abstract
:1. Introduction
2. Results
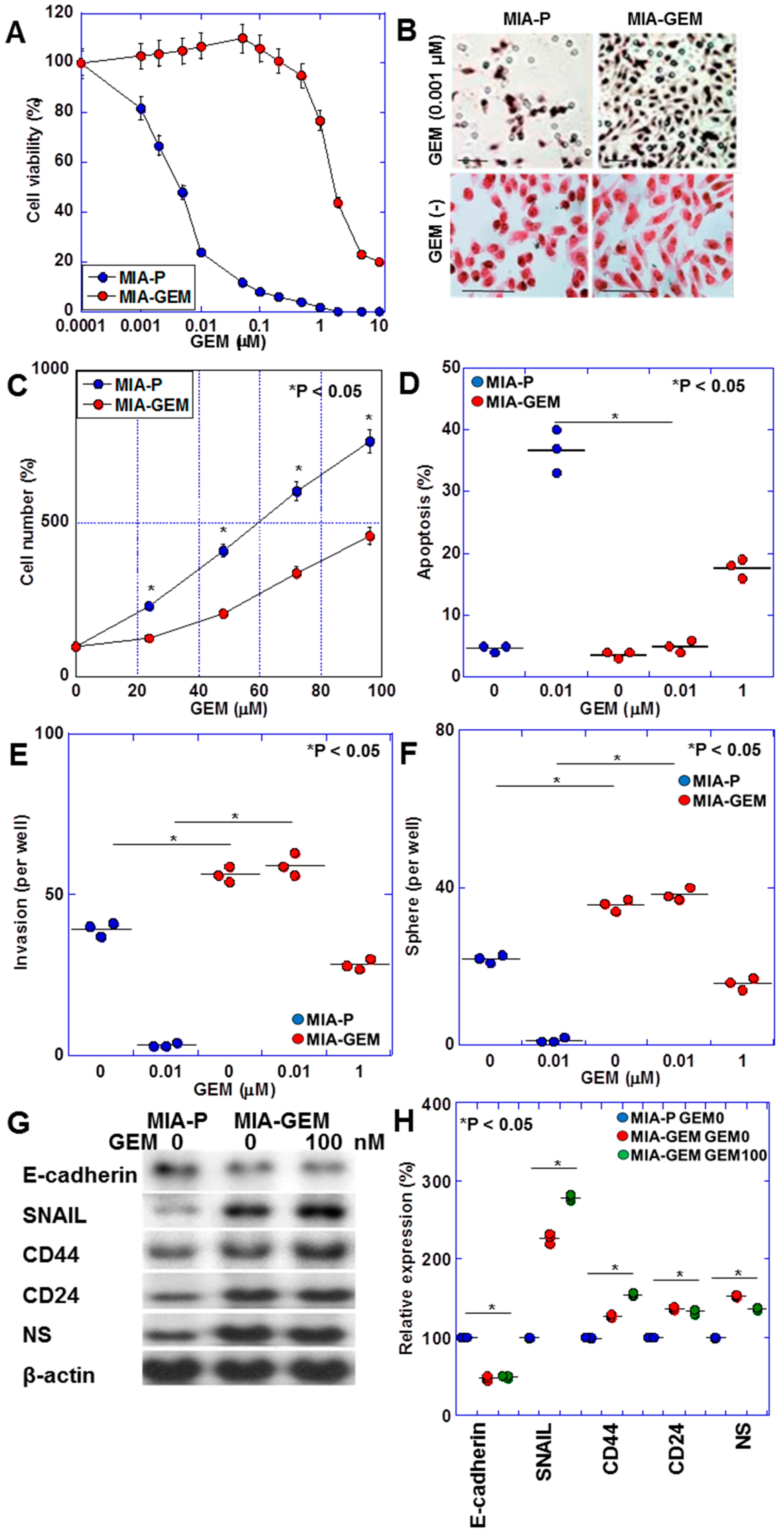
2.1. Establishment of GEM-Resistant PDAC Cells
2.2. Gene Expression Changes in GEM-Resistant PDAC Cells
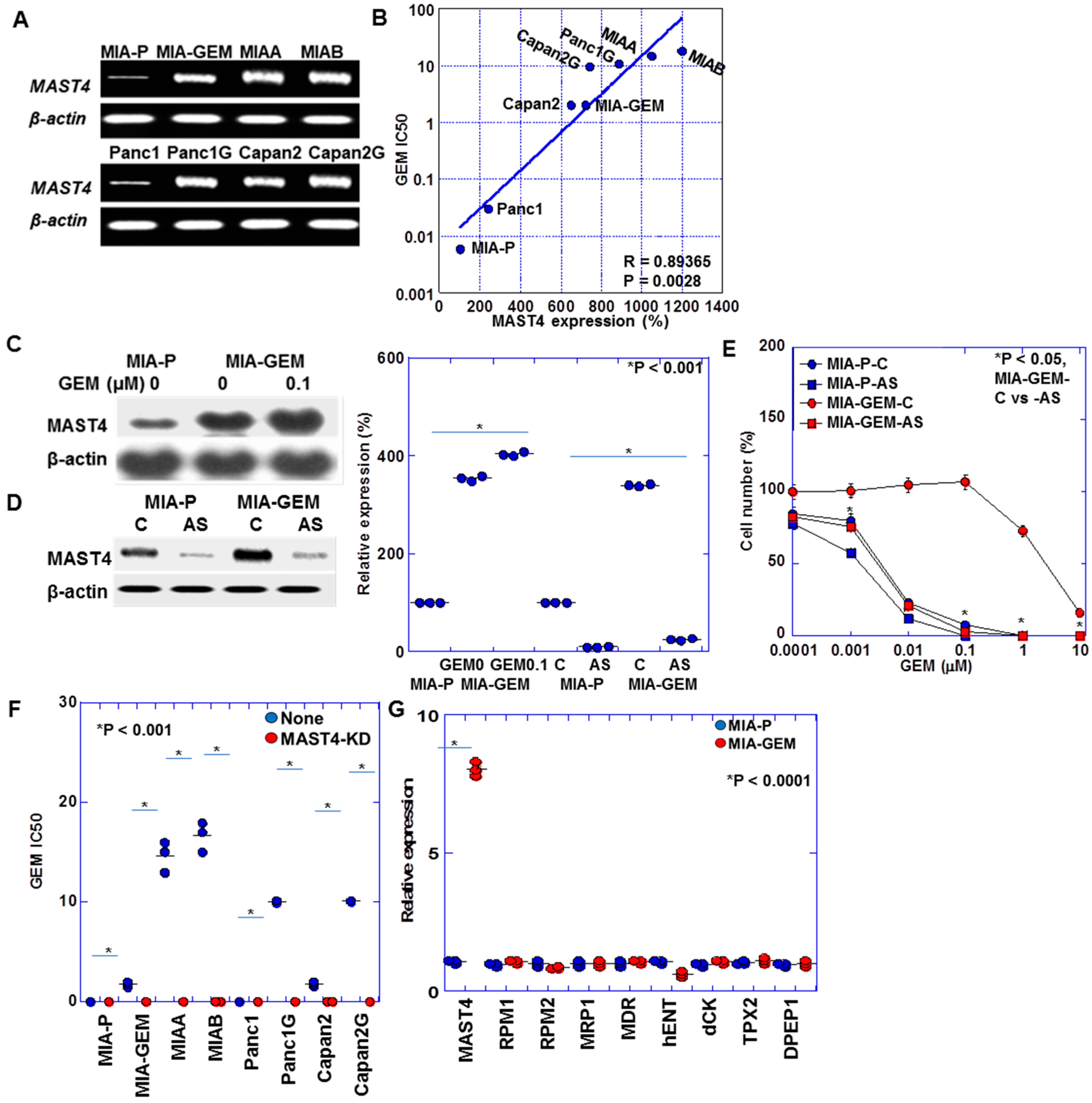
2.3. Identification of MAST4-Related Signals
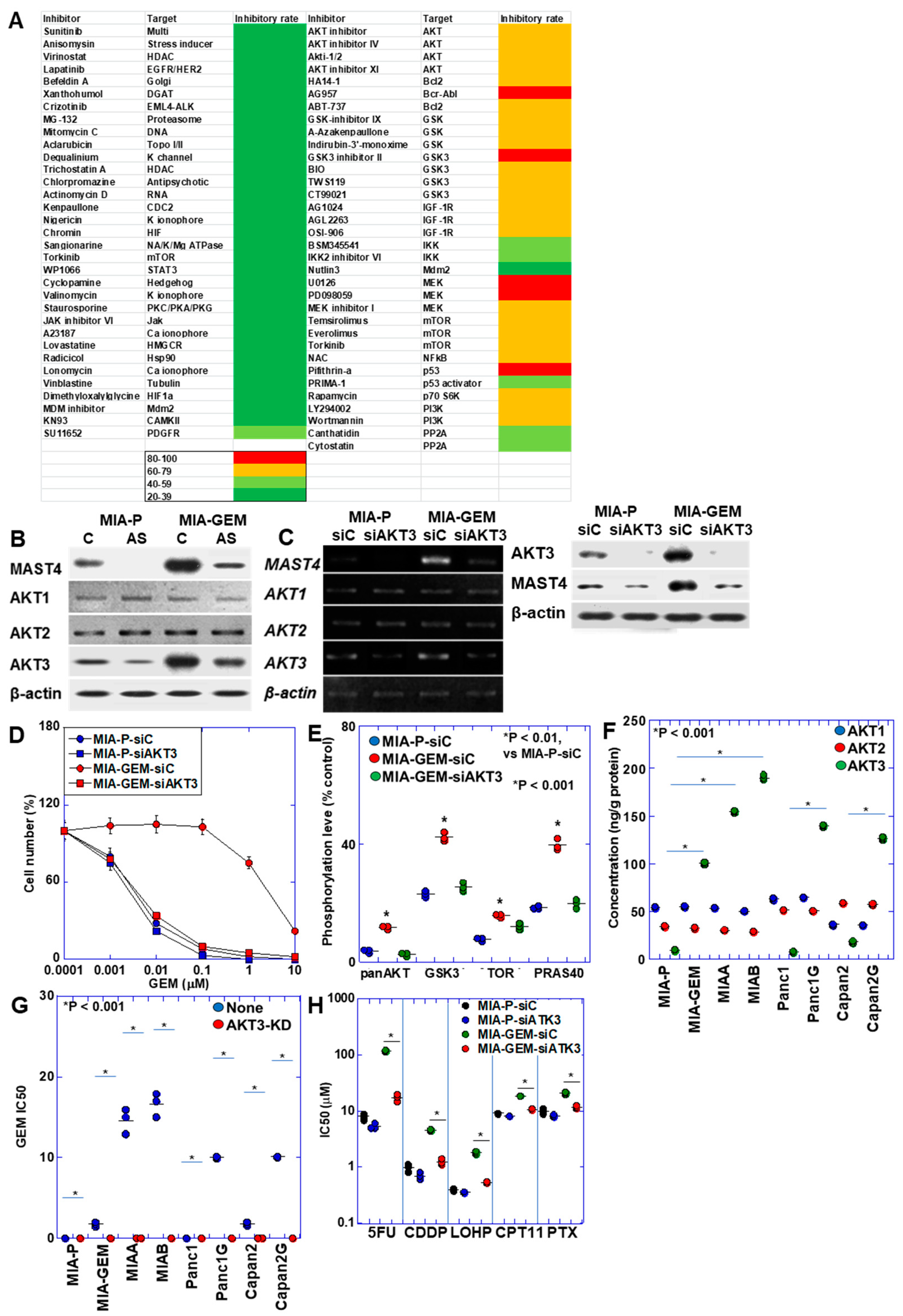
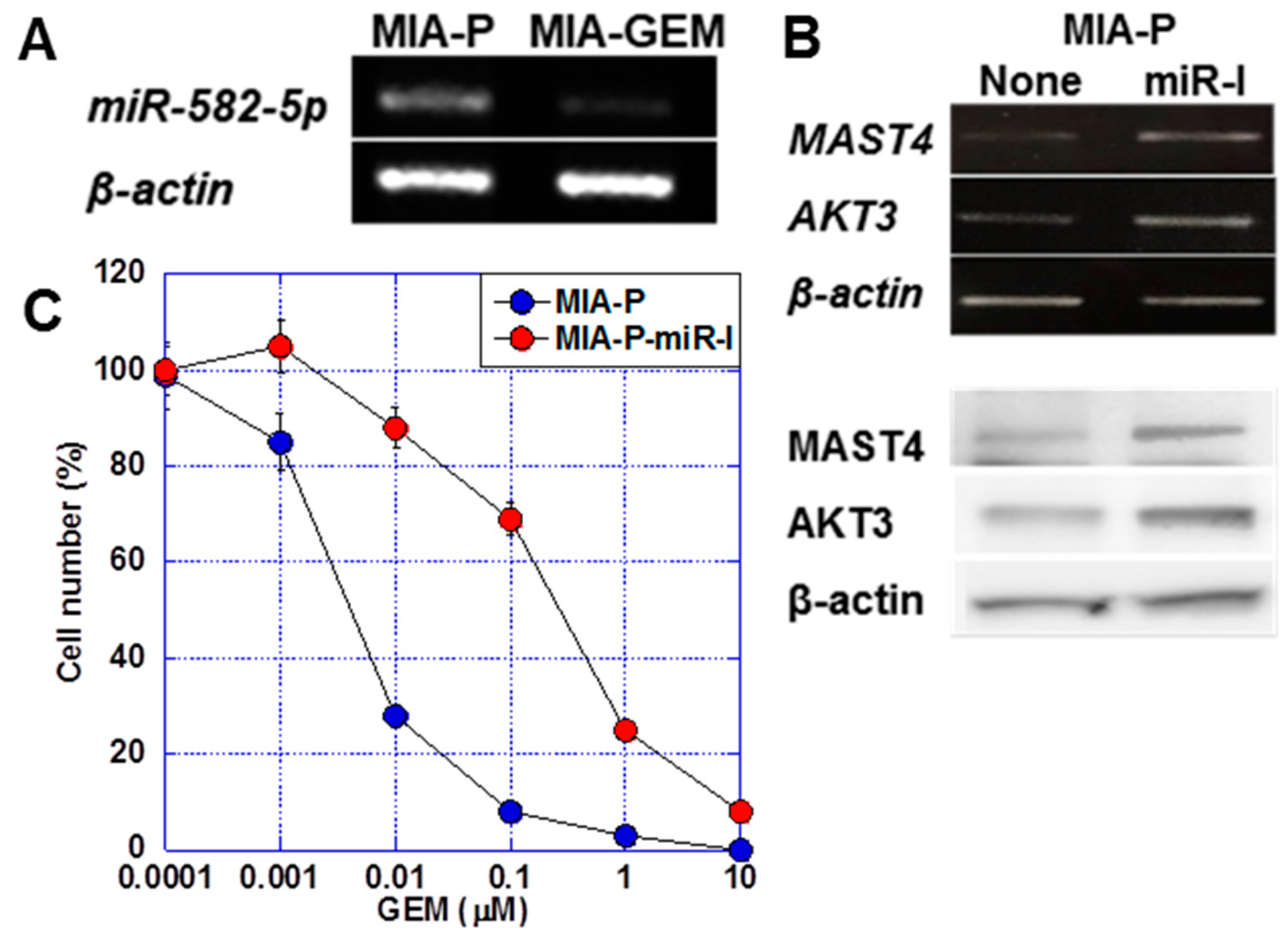
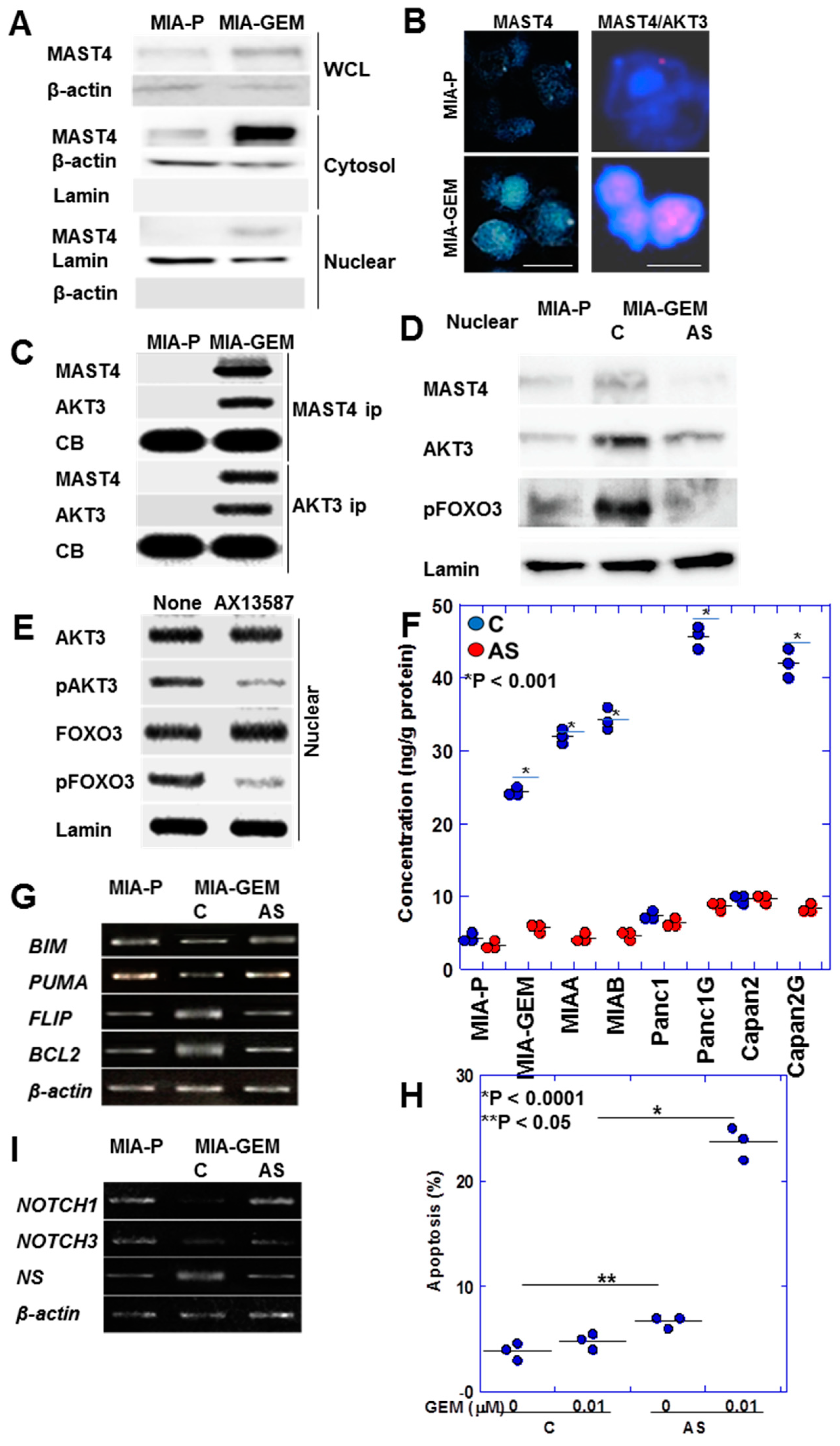
2.4. Regulation of MAST4 and AKT3 Expression
2.5. Nuclear Translocation of MAST4 and AKT3
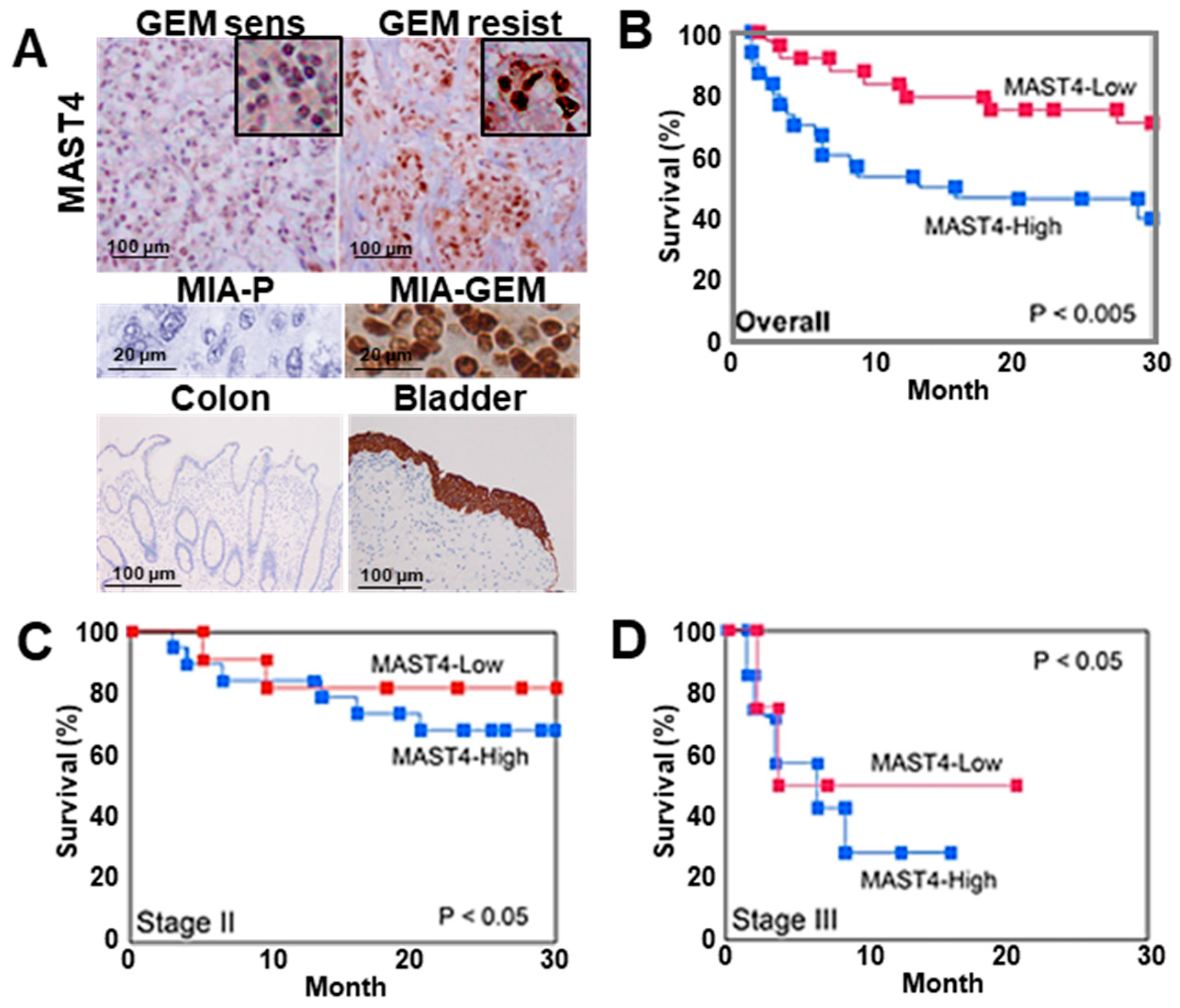
2.6. Role of MAST4 in PDAC Cases
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Growth and Apoptosis
4.3. Sphere Assay
4.4. Chamber Invasion Assay
4.5. Mouse Models
4.6. Antisense Phosphorothioate(S)-Oligodeoxynucleotide Assay
4.7. mRNA Profiling
4.8. Inhibitor Assay
4.9. Western Blotting
4.10. Immunoprecipitation
4.11. Reverse Transcription–Polymerase Chain Reaction (RT–PCR)
4.12. Small Interfering RNA
4.13. Duolink® Proximity Ligation Assay
4.14. Enzyme-Linked Immunosorbent Assay (ELISA) and Fluorometric Assay
4.15. Patients
4.16. Immunohistochemistry
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Higashi, T.; Ishii, T.; Katanoda, K.; Fujishita, M.; Matsuda, T. Cancer Statistics in Japan, 2023; Foundation for Promotion of Cancer Research: Tokyo, Japan, 2013. [Google Scholar]
- Howlander, N.; Noone, A.; Krapcho, M. SEER Cancer Statistics Review, 1975–2017. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 7 November 2023).
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates 2015, 23, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Fujiwara-Tani, R.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Sasaki, T.; Ogata, R.; Ikemoto, A.; Sasaki, R.; Ohmori, H.; et al. Lauric Acid Overcomes Hypoxia-Induced Gemcitabine Chemoresistance in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7506. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara-Tani, R.; Sasaki, T.; Takagi, T.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Gemcitabine Resistance in Pancreatic Ductal Carcinoma Cell Lines Stems from Reprogramming of Energy Metabolism. Int. J. Mol. Sci. 2022, 23, 7824. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.M.; Leung, K.K.; Wheatley, S.C.; Ng, L.J.; Zhou, S.; Ling, K.W.; Sham, M.H.; Koopman, P.; Tam, P.P.; Cheah, K.S. SOX9 directly regulates the type-II collagen gene. Nat. Genet. 1997, 16, 174–178. [Google Scholar] [CrossRef]
- Lee, S.J.; Park, J.; Lee, D.J.; Otsu, K.; Kim, P.; Mizuno, S.; Lee, M.J.; Kim, H.Y.; Harada, H.; Takahashi, S.; et al. Mast4 knockout shows the regulation of spermatogonial stem cell self-renewal via the FGF2/ERM pathway. Cell Death Differ. 2021, 28, 1441–1454. [Google Scholar] [CrossRef]
- Gongol, B.; Marin, T.L.; Jeppson, J.D.; Mayagoitia, K.; Shin, S.; Sanchez, N.; Kirsch, W.M.; Vinters, H.V.; Wilson, C.G.; Ghribi, O.; et al. Cellular hormetic response to 27-hydroxycholesterol promotes neuroprotection through AICD induction of MAST4 abundance and kinase activity. Sci. Rep. 2017, 7, 13898. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, N.; Cao, Y.; Peng, Y.; Lian, A.; Chen, Y.; Wang, P.; Gu, W.; Xiao, B.; Yu, J.; et al. De novo variants in MAST4 related to neurodevelopmental disorders with developmental delay and infantile spasms: Genotype-phenotype association. Front. Mol. Neurosci. 2023, 16, 1097553. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, K.H.; Lee, D.J.; Kim, P.; Park, J.; Kim, S.J.; Jung, H.S. MAST4 controls cell cycle in spermatogonial stem cells. Cell Prolif. 2023, 56, e13390. [Google Scholar] [CrossRef]
- Kim, P.; Park, J.; Lee, D.J.; Mizuno, S.; Shinohara, M.; Hong, C.P.; Jeong, Y.; Yun, R.; Park, H.; Park, S.; et al. Mast4 determines the cell fate of MSCs for bone and cartilage development. Nat. Commun. 2022, 13, 3960. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, F.; Zhang, D.; Huang, J.; Yang, Y.; Xu, J.; Gao, Y.; Ding, H.; Qu, Y.; Zhang, W.; et al. Estrogen-Responsive Gene MAST4 Regulates Myeloma Bone Disease. J. Bone Miner. Res. 2022, 37, 711–723. [Google Scholar] [CrossRef]
- Rodrigues-Ferreira, S.; Morin, M.; Guichaoua, G.; Moindjie, H.; Haykal, M.M.; Collier, O.; Stoven, V.; Nahmias, C. A Network of 17 Microtubule-Related Genes Highlights Functional Deregulations in Breast Cancer. Cancers 2023, 15, 4870. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.L.; Gao, H.; Liu, D.; Philips, T.J.; Ye, Z.; Lee, J.H.; Shi, G.X.; Copenhaver, K.; Zhang, L.; Wei, L.; et al. Glucocorticoids unmask silent non-coding genetic risk variants for common diseases. Nucleic Acids Res. 2022, 50, 11635–11653. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Gullì, M.; Facchinetti, R.; Minacori, M.; Mancinelli, R.; Percaccio, E.; Scuderi, C.; Eufemi, M.; Di Sotto, A. Sorafenib Chemosensitization by Caryophyllane Sesquiterpenes in Liver, Biliary, and Pancreatic Cancer Cells: The Role of STAT3/ABC Transporter Axis. Pharmaceutics 2022, 14, 1264. [Google Scholar] [CrossRef]
- Cao, J.; Yang, J.; Ramachandran, V.; Arumugam, T.; Deng, D.; Li, Z.; Xu, L.; Logsdon, C.D. TM4SF1 Promotes Gemcitabine Resistance of Pancreatic Cancer In Vitro and In Vivo. PLoS ONE 2015, 10, e0144969. [Google Scholar] [CrossRef]
- Sierzega, M.; Pach, R.; Kulig, P.; Legutko, J.; Kulig, J. Prognostic Implications of Expression Profiling for Gemcitabine-Related Genes (hENT1, dCK, RRM1, RRM2) in Patients with Resectable Pancreatic Adenocarcinoma Receiving Adjuvant Chemotherapy. Pancreas 2017, 46, 684–689. [Google Scholar] [CrossRef]
- Guenther, M.; Surendran, S.A.; Haas, M.; Heinemann, V.; von Bergwelt-Baildon, M.; Engel, J.; Werner, J.; Boeck, S.; Ormanns, S. TPX2 expression as a negative predictor of gemcitabine efficacy in pancreatic cancer. Br. J. Cancer 2023, 129, 175–182. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, W.; Ran, Y.; Xiong, Y.; Zhong, Z.; Fan, X.; Wang, Z.; Ye, Q. miR-582-5p inhibits proliferation of hepatocellular carcinoma by targeting CDK1 and AKT3. Tumour Biol. 2015, 36, 8309–8316. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tao, L.P.; Yao, S.C.; Huang, Q.K.; Chen, Z.F.; Sun, Y.J.; Jin, S.Q. MicroRNA-582-5p suppressed gastric cancer cell proliferation via targeting AKT3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5112–5120. [Google Scholar] [PubMed]
- Li, B.; Cociorva, O.M.; Nomanbhoy, T.; Weissig, H.; Li, Q.; Nakamura, K.; Liyanage, M.; Zhang, M.C.; Shih, A.Y.; Aban, A.; et al. Hit-to-lead optimization and kinase selectivity of imidazo[1,2-a]quinoxalin-4-amine derived JNK1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5217–5222. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.D.; Webb, A.E.; Brunet, A.; Rando, T.A. FOXO3 promotes quiescence in adult muscle stem cells during the process of self-renewal. Stem Cell Rep. 2014, 2, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Tamase, A.; Muraguchi, T.; Naka, K.; Tanaka, S.; Kinoshita, M.; Hoshii, T.; Ohmura, M.; Shugo, H.; Ooshio, T.; Nakada, M.; et al. Identification of tumor-initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc. Natl. Acad. Sci. USA 2009, 106, 17163–17168. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Human Protein Atlas—MAST4. Science 2015, 347, 1260419. Available online: https://v20.proteinatlas.org/search/MAST4 (accessed on 4 March 2018). [CrossRef]
- Missiroli, S.; Etro, D.; Buontempo, F.; Ye, K.; Capitani, S.; Neri, L.M. Nuclear translocation of active AKT is required for erythroid differentiation in erythropoietin treated K562 erythroleukemia cells. Int. J. Biochem. Cell Biol. 2009, 41, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Xuan Nguyen, T.L.; Choi, J.W.; Lee, S.B.; Ye, K.; Woo, S.D.; Lee, K.H.; Ahn, J.Y. Akt phosphorylation is essential for nuclear translocation and retention in NGF-stimulated PC12 cells. Biochem. Biophys. Res. Commun. 2006, 349, 789–798. [Google Scholar] [CrossRef]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef]
- Jain, M.V.; Jangamreddy, J.R.; Grabarek, J.; Schweizer, F.; Klonisch, T.; Cieślar-Pobuda, A.; Łos, M.J. Nuclear localized Akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1). Cell Cycle 2015, 14, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Nakazawa, T.; Nakano, I.; Mori, N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 2000, 349 Pt 2, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Maatz, H.; Kim, H.S.; Yang, J.; Abid, M.R.; Aird, W.C.; Walsh, K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J. Biol. Chem. 2004, 279, 1513–1525. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Yin, Y.; Li, M.; Wang, B.; Yang, L.; Jiang, Y. FOXO3-mediated up-regulation of Bim contributes to rhein-induced cancer cell apoptosis. Apoptosis 2015, 20, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, J.; Tang, J.; Liu, X.; Zhong, Q.; Wang, F.; Hu, W.; Yuan, Z.; Nie, C.; Wei, Y. JNK- and Akt-mediated Puma expression in the apoptosis of cisplatin-resistant ovarian cancer cells. Biochem. J. 2012, 444, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Maeda, Y.; Kita, K.; Murakami, K.; Saya, H.; Takemura, H.; Inaki, N.; Oshima, M.; Oshima, H. FOXO3 is a latent tumor suppressor for FOXO3-positive and cytoplasmic-type gastric cancer cells. Oncogene 2021, 40, 3072–3086. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Chen, Y.T.; Wu, C.L.; Yu, C.P. Expression of p-FOXO3/FOXO3 in bladder cancer and its correlation with clinicopathology and tumor recurrence. Int. J. Clin. Exp. Pathol. 2017, 10, 11069–11074. [Google Scholar] [PubMed]
- Liu, H.; Song, Y.; Qiu, H.; Liu, Y.; Luo, K.; Yi, Y.; Jiang, G.; Lu, M.; Zhang, Z.; Yin, J.; et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 2020, 27, 966–983. [Google Scholar] [CrossRef] [PubMed]
- Audesse, A.J.; Karashchuk, G.; Gardell, Z.A.; Lakis, N.S.; Maybury-Lewis, S.Y.; Brown, A.K.; Leeman, D.S.; Teo, Y.V.; Neretti, N.; Anthony, D.C.; et al. FOXO3 regulates a common genomic program in aging and glioblastoma stem cells. Aging Cancer 2021, 2, 137–159. [Google Scholar] [CrossRef]
- Garland, P.; Quraishe, S.; French, P.; O’Connor, V. Expression of the MAST family of serine/threonine kinases. Brain Res. 2008, 1195, 12–19. [Google Scholar] [CrossRef]
- Songyang, Z.; Fanning, A.S.; Fu, C.; Xu, J.; Marfatia, S.M.; Chishti, A.H.; Crompton, A.; Chan, A.C.; Anderson, J.M.; Cantley, L.C. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 1997, 275, 73–77. [Google Scholar] [CrossRef]
- Hung, A.Y.; Sheng, M. PDZ domains: Structural modules for protein complex assembly. J. Biol. Chem. 2002, 277, 5699–5702. [Google Scholar] [CrossRef]
- Jeleń, F.; Oleksy, A.; Smietana, K.; Otlewski, J. PDZ domains—Common players in the cell signaling. Acta Biochim. Pol. 2003, 50, 985–1017. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.Z.; Lim, W.A. Mechanism and role of PDZ domains in signaling complex assembly. J. Cell Sci. 2001, 114 Pt 18, 3219–3231. [Google Scholar] [CrossRef]
- Valiente, M.; Andrés-Pons, A.; Gomar, B.; Torres, J.; Gil, A.; Tapparel, C.; Antonarakis, S.E.; Pulido, R. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J. Biol. Chem. 2005, 280, 28936–28943. [Google Scholar] [CrossRef]
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Préhaud, C.; Wolff, N.; Terrien, E.; Lafage, M.; Mégret, F.; Babault, N.; Cordier, F.; Tan, G.S.; Maitrepierre, E.; Ménager, P.; et al. Attenuation of rabies virulence: Takeover by the cytoplasmic domain of its envelope protein. Sci. Signal 2010, 3, ra5. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Kishi, S.; Fujiwara-Tani, R.; Honoki, K.; Sasaki, R.; Mori, S.; Ohmori, H.; Sasaki, T.; Miyagawa, Y.; Kawahara, I.; Kido, A.; et al. Oxidized high mobility group B-1 enhances metastability of colorectal cancer via modification of mesenchymal stem/stromal cells. Cancer Sci. 2022, 113, 2904–2915. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Luo, Y.; Fujii, K.; Sasahira, T.; Moriwaka, Y.; Tatsumoto, N.; Sasaki, T.; Yamashita, Y.; Ohmori, H. CD10 enhances metastasis of colorectal cancer by abrogating the anti-tumoural effect of methionine-enkephalin in the liver. Gut 2010, 59, 348–356. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Yasui, W.; Pettaway, C.A.; Yano, S.; Oue, N.; Tahara, E.; Fidler, I.J. Interferon-alpha prevents selection of doxorubicin-resistant undifferentiated-androgen-insensitive metastatic human prostate cancer cells. Prostate 2001, 49, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y. Investigation of the freely available easy-to-use software ’EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Increased | Fold Change (MIA-P = 1) |
|---|---|
| MAST4 | 7.1782656 |
| GJA1 | 6.209089 |
| PDE1A | 6.1802254 |
| FLI1 | 6.1545568 |
| ELOVL6 | 6.0397415 |
| MMP1 | 5.802817 |
| SYTL4 | 5.648421 |
| ADD3 | 5.6095138 |
| RHOBTB3 | 5.5603056 |
| MAP3K5 | 5.4986305 |
| Decreased | Fold Change (MIA-P = 1) |
| IFI16 | −8.182869 |
| ATPBD4 | −5.3832583 |
| PTPRZ1 | −5.1480074 |
| ACSL6 | -5.0153513 |
| AQP4 | −4.8476763 |
| COL1A1 | −4.837364 |
| GIT2 | −4.766745 |
| LOC644242 | −4.5996995 |
| LPHN3 | −4.380941 |
| SFRP1 | −4.3634024 |
| WDR26 | −4.1247654 |
| Parameter | Nuclear MAST4 | |||
|---|---|---|---|---|
| n | Index (%) 1 | p-Value 2 | ||
| Sex | Male | 54 | 45 ± 15 | |
| Female | 37 | 54 ± 20 | NS | |
| Age | 23–52 y | 46 | 48 ± 18 | |
| 52–78 y | 45 | 49 ± 17 | NS | |
| PS 3 | 1 | 32 | 48 ± 19 | |
| 2 | 59 | 49 ± 22 | NS | |
| Grade 4 | G1 | 31 | 28 ± 12 | |
| G2 | 29 | 48 ± 18 | ||
| G3 | 31 | 68 ± 13 | <0.0001 | |
| T factor 4 | pT1-2 | 28 | 37 ± 14 | |
| pT3-4 | 63 | 54 ± 19 | <0.0001 | |
| N factor 4 | pN0 | 79 | 46 ± 18 | |
| pN1-2 | 12 | 68 ± 21 | 0.0002 | |
| Stage 4 | I | 26 | 17 ± 14 | |
| II | 51 | 24 ± 18 | ||
| III–IV | 14 | 73 ± 23 | <0.0001 | |
| RECIST 5 | CR | 2 | 15 ± 18 | |
| PR | 15 | 18 ± 14 | <0.0001 | |
| SD | 10 | 26 ± 15 | ||
| PD | 64 | 92 ± 17 | <0.0001 | |
| GEM alone 6 | CR/PR/SD | 21 | 23 ± 13 | |
| PD | 22 | 92 ± 16 | <0.0001 |
| Parameter | MAST4-H 1 | MAST4-L 1 | p-Value 2 | |
|---|---|---|---|---|
| n | 14 | 13 | ||
| Nuclear MAST4 (%) | 68 (48–81) | 37 (14–47) | <0.0001 | |
| Sex | Male | 7 | 7 | |
| Female | 7 | 6 | NS | |
| Age | <52 y | 5 | 5 | |
| <53 y | 9 | 8 | NS | |
| PS 3 | 1 | 6 | 6 | |
| 2 | 8 | 7 | NS | |
| Grade 4 | G1 | 4 | 4 | |
| G2 | 5 | 5 | ||
| G3 | 5 | 4 | NS | |
| T factor 4 | pT1-2 | 12 | 11 | |
| pT3-4 | 2 | 2 | NS | |
| N factor 4 | pN0 | 8 | 6 | |
| pN1-2 | 6 | 7 | NS | |
| Stage 4 | I | 0 | 1 | |
| II | 9 | 7 | ||
| III–IV | 5 | 5 | NS | |
| RECIST 5 | CR | 0 | 0 | |
| PR | 0 | 3 | ||
| SD | 3 | 6 | ||
| PD | 11 | 4 | 0.0268 | |
| Parameters | Hazard Ratio | 95% Confidential Interval | p-Value |
|---|---|---|---|
| T factor | 0.06668 | 0.005297–0.8393 | 0.03612 |
| N factor | 0.18460 | 0.024960–1.3650 | 0.09786 |
| Stage | 4.24400 | 1.159000–15.5400 | 0.02904 |
| MAST4 | 91.06000 | 2.837000–2923.0000 | 0.01080 |
| Primer Set | |||
|---|---|---|---|
| Gene Symbol | Gene Bank ID | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
| MAST4 | NM_015183.2 | ttccccaactggaatctgag | aggtggtgcttttggttttg |
| AKT1 | KR710120.1 | gcaccttccatgtggagact | cccagcagcttcaggtactc |
| AKT2 | M95936.1 | gaggtcatggagcacaggtt | ctggtccagctccagtaagc |
| AKT3 | AJ245709.1 | cagtagactggtggggccta | atcaagagccctgaaagcaa |
| BIM | AY352518.1 | gcccctacctccctacagac | atggtggtggccatacaaat |
| PUMA | AF354654.1 | ggagcagcacctggagtc | tactgtgcgttgaggtcgtc |
| FLIP | AB038972.2 | cttggccaatttgcctgtat | tctttggcttccctgctaga |
| BCL2 | M13994.1 | acgacaaccgggagatagtg | catcccagcctccgttatcc |
| NOTCH1 | CR457221.1 | gatgtgtggactgtggcact | tgtgttgctggagcatcttc |
| NOTCH3 | U97669.1 | tgtggacgagtgctctatcg | aatgtccacctcgcaatagg |
| β-actin | NM_001101.3 | ggacttcgagcaagagatgg | agcactgtgttggcgtacag |
| miR-582-5p | NR_030308.1 | tgtgctctttgattacagttgttc | caccctttgggttcagttgt |
| Antisense | |||
| MAST4 | AS | aatgctggactcatccat | |
| Control | nnnnnnnnnnnnnnnnnn | ||
| Antibody | |||
| Protein | Clone | Company | |
| E-cadherin | EP700Y | Abcam, Cambridge, MA, USA | |
| SNAIL | L70G2 | Biocompare. South San Francisco, CA, USA | |
| CD44 | F-4 | Santa Cruz, Santa Cruz, CA, USA | |
| CD24 | ERP19925 | Abcam, Cambridge, MA, USA | |
| NS | - | antibodies-online GmbH, Aachen, Germany | |
| MAST4 | - | Merck, Tokyo, Japan | |
| AKT1 | 1F7E10 | Abcam, Cambridge, MA, USA | |
| AKT2 | EP1676 | Abcam, Cambridge, MA, USA | |
| AKT3 | EE-M14 | Santa Cruz, Santa Cruz, CA, USA | |
| Lamin | - | Abcam, Cambridge, MA, USA | |
| FOXO3 | - | Abcam, Cambridge, MA, USA | |
| pAKT3 (pY312) | - | Bio-Rad. Hercules, CA, USA | |
| pFOXO3 (pS322/S325) | - | Cusabio, Houston, TX, USA | |
| β-actin | - | Abcam, Cambridge, MA, USA | |
| ELISA | |||
| Protein | Catalog# | ||
| AKT1 | LS-F68359 | LS Bio, Shirley, MA, USA | |
| AKT2 | ab208986 | Abcam, Cambridge, MA, USA | |
| AKT3 | EH6215 | Fine Test, Boulder CO, USA | |
| pFoxO3 | 28755-1-AP | Proteintech, Rosemont, IL, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiwara-Tani, R.; Sasaki, T.; Bhawal, U.K.; Mori, S.; Ogata, R.; Sasaki, R.; Ikemoto, A.; Kishi, S.; Fujii, K.; Ohmori, H.; et al. Nuclear MAST4 Suppresses FOXO3 through Interaction with AKT3 and Induces Chemoresistance in Pancreatic Ductal Carcinoma. Int. J. Mol. Sci. 2024, 25, 4056. https://doi.org/10.3390/ijms25074056
Fujiwara-Tani R, Sasaki T, Bhawal UK, Mori S, Ogata R, Sasaki R, Ikemoto A, Kishi S, Fujii K, Ohmori H, et al. Nuclear MAST4 Suppresses FOXO3 through Interaction with AKT3 and Induces Chemoresistance in Pancreatic Ductal Carcinoma. International Journal of Molecular Sciences. 2024; 25(7):4056. https://doi.org/10.3390/ijms25074056
Chicago/Turabian StyleFujiwara-Tani, Rina, Takamitsu Sasaki, Ujjal Kumar Bhawal, Shiori Mori, Ruiko Ogata, Rika Sasaki, Ayaka Ikemoto, Shingo Kishi, Kiyomu Fujii, Hitoshi Ohmori, and et al. 2024. "Nuclear MAST4 Suppresses FOXO3 through Interaction with AKT3 and Induces Chemoresistance in Pancreatic Ductal Carcinoma" International Journal of Molecular Sciences 25, no. 7: 4056. https://doi.org/10.3390/ijms25074056
APA StyleFujiwara-Tani, R., Sasaki, T., Bhawal, U. K., Mori, S., Ogata, R., Sasaki, R., Ikemoto, A., Kishi, S., Fujii, K., Ohmori, H., Sho, M., & Kuniyasu, H. (2024). Nuclear MAST4 Suppresses FOXO3 through Interaction with AKT3 and Induces Chemoresistance in Pancreatic Ductal Carcinoma. International Journal of Molecular Sciences, 25(7), 4056. https://doi.org/10.3390/ijms25074056