
Isothermal Nucleic Acid Amplification-Based Lateral Flow Testing for the Detection of Plant Viruses


Abstract
:1. Introduction
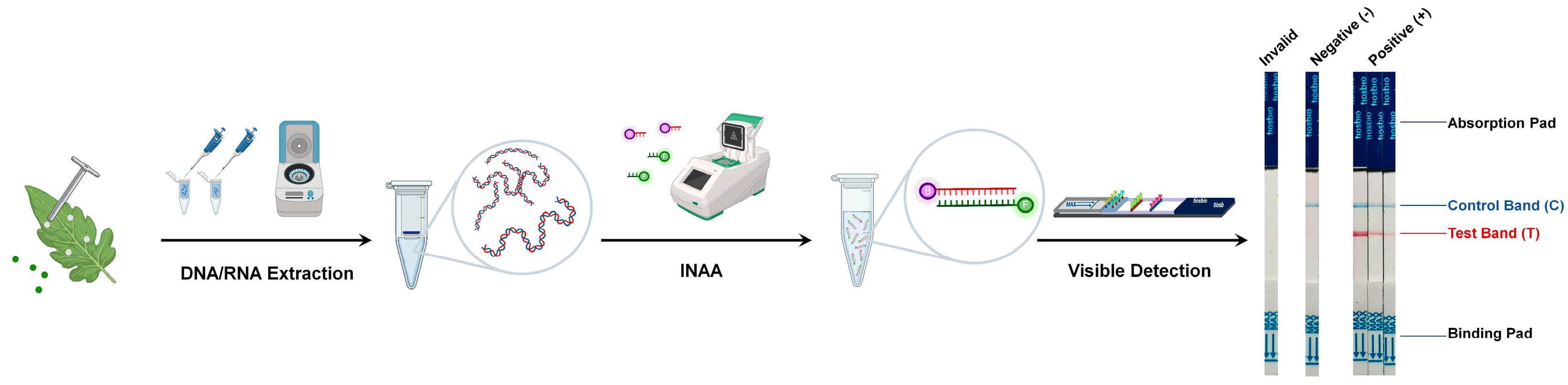
2. INAA-LFT Workflow
2.1. Amplification of the Target Nucleic Acid Fragments from Pathogens
2.2. Labeling of the Target Nucleic Acid Fragments from Pathogens
2.3. Visible Detection of Target Nucleic Acid Fragments from Pathogens
3. Application of INAA-LFT in the Detection of Plant Viruses
3.1. LAMP-LFT Detection of Plant Viruses
3.2. RPA-LFT Detection of Plant Viruses
3.3. RAA-LFT Detection of Plant Viruses
3.4. CRISPR-CAS System-Integrated LFT Detection of Plant Viruses
4. Factors Influencing the Implementation of INAA-LFT for the On-Site Detection of Plant Viruses
4.1. Sensitivity and Specificity
4.2. Detection Duration
4.3. Ease of Operation
5. The Future Trajectory of INAA-LFT in Detection of Plant Virus
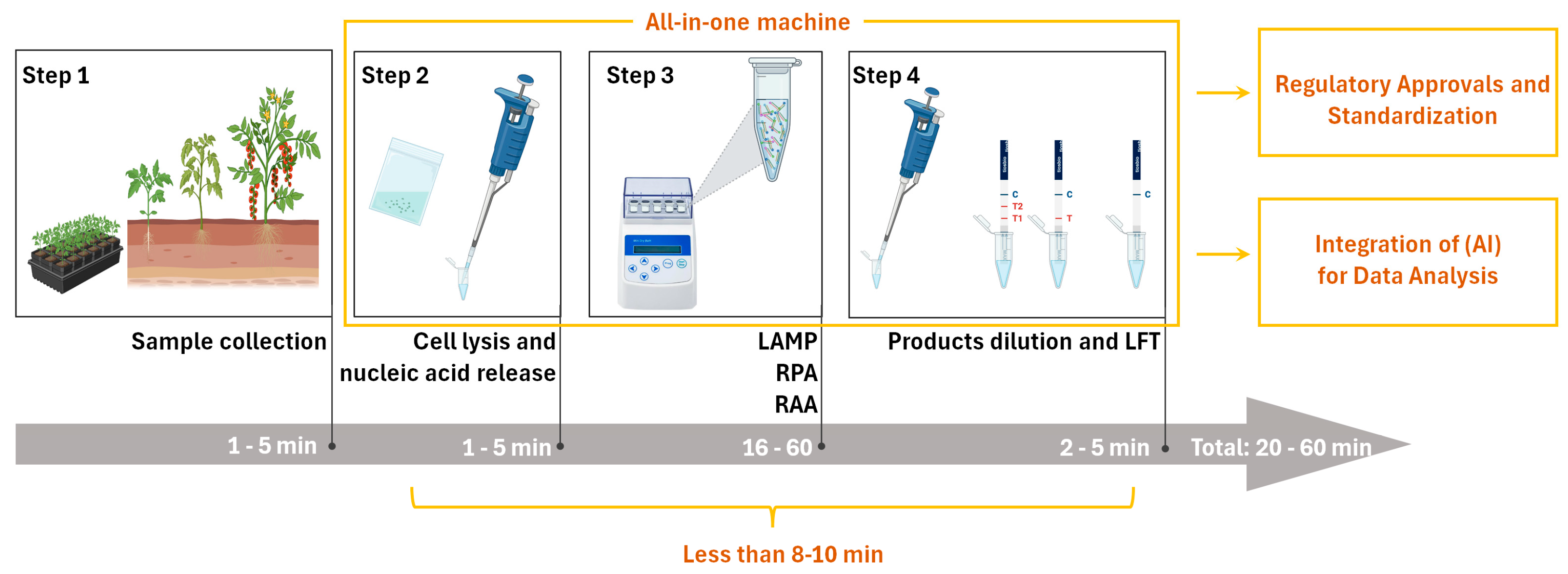
5.1. Expedited Detecting Duration
5.2. Operational Simplification and Automation
5.3. Portable Devices
5.4. Regulatory Approvals and Standardization
5.5. Integration of Artificial Intelligence (AI) for Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhat, A.I.; Aman, R.; Mahfouz, M. Onsite detection of plant viruses using isothermal amplification assays. Plant Biotechnol. J. 2022, 20, 1859–1873. [Google Scholar] [CrossRef]
- Wong, Y.P.; Othman, S.; Lau, Y.L.; Radu, S.; Chee, H.Y. Loop-mediated isothermal amplification (LAMP): A versatile technique for detection of micro-organisms. J. Appl. Microbiol. 2018, 124, 626–643. [Google Scholar] [CrossRef]
- Boonham, N.; Kreuze, J.; Winter, S.; van der Vlugt, R.; Bergervoet, J.; Tomlinson, J.; Mumford, R. Methods in virus diagnostics: From ELISA to next generation sequencing. Virus Res. 2014, 186, 20–31. [Google Scholar] [CrossRef]
- Liu, Y.; Zhan, L.; Qin, Z.; Sackrison, J.; Bischof, J.C. Ultrasensitive and Highly Specific Lateral Flow Assays for Point-of-Care Diagnosis. ACS Nano 2021, 15, 3593–3611. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Safenkova, I.V.; Zherdev, A.V.; Dzantiev, B.B. The Potential Use of Isothermal Amplification Assays for In-Field Diagnostics of Plant Pathogens. Plants 2021, 10, 2424. [Google Scholar] [CrossRef]
- Lau, H.Y.; Botella, J.R. Advanced DNA-Based Point-of-Care Diagnostic Methods for Plant Diseases Detection. Front. Plant Sci. 2017, 8, 2016. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Safenkova, I.V.; Zherdev, A.V.; Dzantiev, B.B. Recombinase Polymerase Amplification Assay with and without Nuclease-Dependent-Labeled Oligonucleotide Probe. Int. J. Mol. Sci. 2021, 22, 11885. [Google Scholar] [CrossRef]
- Sang, P.; Hu, Z.; Cheng, Y.; Yu, H.; Xie, Y.; Yao, W.; Guo, Y.; Qian, H. Nucleic Acid Amplification Techniques in Immunoassay: An Integrated Approach with Hybrid Performance. J. Agric. Food Chem. 2021, 69, 5783–5797. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef]
- Mori, Y.; Notomi, T. Loop-mediated isothermal amplification (LAMP): A rapid, accurate, and cost-effective diagnostic method for infectious diseases. J. Infect. Chemother. 2009, 15, 62–69. [Google Scholar] [CrossRef]
- Gill, P.; Ghaemi, A. Nucleic acid isothermal amplification technologies: A review. Nucleosides Nucleotides Nucleic Acids 2008, 27, 224–243. [Google Scholar] [CrossRef]
- Tomlinson, J.A.; Ostoja-Starzewska, S.; Adams, I.P.; Miano, D.W.; Abidrabo, P.; Kinyua, Z.; Alicai, T.; Dickinson, M.J.; Peters, D.; Boonham, N.; et al. Loop-mediated isothermal amplification for rapid detection of the causal agents of cassava brown streak disease. J. Virol. Methods 2013, 191, 148–154. [Google Scholar] [CrossRef]
- Edgu, G.; Freund, L.J.; Hartje, S.; Tacke, E.; Hofferbert, H.R.; Twyman, R.M.; Noll, G.A.; Muth, J.; Prufer, D. Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification. Int. J. Mol. Sci. 2020, 21, 8741. [Google Scholar] [CrossRef]
- Lu, H.; Tang, J.; Sun, K.; Yu, X. Identification of a New Badnavirus in the Chinaberry (Melia azedarach) Tree and Establishment of a LAMP-LFD Assay for Its Rapid and Visual Detection. Viruses 2021, 13, 2408. [Google Scholar] [CrossRef]
- Kim, J.; Easley, C.J. Isothermal DNA amplification in bioanalysis: Strategies and applications. Bioanalysis 2011, 3, 227–239. [Google Scholar] [CrossRef]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef]
- Deng, H.; Gao, Z. Bioanalytical applications of isothermal nucleic acid amplification techniques. Anal. Chim. Acta 2015, 853, 30–45. [Google Scholar] [CrossRef]
- Zhang, S.; Ravelonandro, M.; Russell, P.; McOwen, N.; Briard, P.; Bohannon, S.; Vrient, A. Rapid diagnostic detection of plum pox virus in Prunus plants by isothermal AmplifyRP((R)) using reverse transcription-recombinase polymerase amplification. J. Virol. Methods 2014, 207, 114–120. [Google Scholar] [CrossRef]
- Zhao, C.; Sun, F.; Li, X.; Lan, Y.; Du, L.; Zhou, T.; Zhou, Y. Reverse transcription-recombinase polymerase amplification combined with lateral flow strip for detection of rice black-streaked dwarf virus in plants. J. Virol. Methods 2019, 263, 96–100. [Google Scholar] [CrossRef]
- Cao, Y.; Yan, D.; Wu, X.; Chen, Z.; Lai, Y.; Lv, L.; Yan, F.; Chen, J.; Zheng, H.; Song, X. Rapid and visual detection of milk vetch dwarf virus using recombinase polymerase amplification combined with lateral flow strips. Virol. J. 2020, 17, 102. [Google Scholar] [CrossRef]
- Kim, D.H.; Jeong, R.D.; Choi, S.; Ju, H.J.; Yoon, J.Y. Application of Rapid and Reliable Detection of Cymbidium Mosaic Virus by Reverse Transcription Recombinase Polymerase Amplification Combined with Lateral Flow Immunoassay. Plant Pathol. J. 2022, 38, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Lee, H.J.; Kim, S.M.; Jeong, R.D. Rapid and Visual Detection of Barley Yellow Dwarf Virus by Reverse Transcription Recombinase Polymerase Amplification with Lateral Flow Strips. Plant Pathol. J. 2022, 38, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Q.; Zhao, X.X.; Wang, D.; Zhang, P.J.; Hu, X.N.; Wei, S.; Liu, J.Y.; Ye, Z.H.; Yu, X.P. A reverse transcription-cross-priming amplification method with lateral flow dipstick assay for the rapid detection of Bean pod mottle virus. Sci. Rep. 2022, 12, 681. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, H.Y.; Jiang, D.M.; Liu, M.; Zhang, W.; Yan, J.Y. A rapid detection of tomato yellow leaf curl virus using recombinase polymerase amplification-lateral flow dipstick assay. Lett. Appl. Microbiol. 2022, 74, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Greeshma, M.; Bhat, A.I.; Jeevalatha, A. Rapid onsite detection of piper yellow mottle virus infecting black pepper by recombinase polymerase amplification-lateral flow assay (RPA-LFA). J. Virol. Methods 2023, 315, 114695. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.; Zhang, Z.; Zhang, Y.; Li, P.; Chen, X.; Liu, M.; Lu, Q.; Li, Z.; Wei, Z.; et al. Development of Recombinase Polymerase Amplification Combined with Lateral Flow Strips for Rapid Detection of Cowpea Mild Mottle Virus. Plant Pathol. J. 2023, 39, 486–493. [Google Scholar] [CrossRef]
- Yilmaz, S.; Batuman, O. Development of a reverse transcription recombinase polymerase amplification combined with lateral flow assay for equipment-free on-site field detection of tomato chlorotic spot virus. Virol. J. 2023, 20, 136. [Google Scholar] [CrossRef]
- Zhang, A.L.; Shi, X.; Xie, C.; Yu, F.; Gao, Z.; Xu, Y.; Liu, Z. Rapid and Visual Detection of Actinidia Chlorotic Ringspot-Associated Virus Using One-Step Reverse-Transcription Recombinase Polymerase Amplification Combined with Lateral Flow Dipstick Assay. Plant Dis. 2023, 107, 3701–3707. [Google Scholar] [CrossRef]
- Hammond, R.W.; Zhang, S. Development of a rapid diagnostic assay for the detection of tomato chlorotic dwarf viroid based on isothermal reverse-transcription-recombinase polymerase amplification. J. Virol. Methods 2016, 236, 62–67. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Kokane, S.B.; Gowda, S. Development of a reverse transcription recombinase polymerase based isothermal amplification coupled with lateral flow immunochromatographic assay (CTV-RT-RPA-LFICA) for rapid detection of Citrus tristeza virus. Sci. Rep. 2020, 10, 20593. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, R.; Zhang, H.; Wang, J.; Lu, Y.; Zhang, D.; Yang, L. PAM-free loop-mediated isothermal amplification coupled with CRISPR/Cas12a cleavage (Cas-PfLAMP) for rapid detection of rice pathogens. Biosens. Bioelectron. 2022, 204, 114076. [Google Scholar] [CrossRef]
- Mekuria, T.A.; Zhang, S.; Eastwell, K.C. Rapid and sensitive detection of Little cherry virus 2 using isothermal reverse transcription-recombinase polymerase amplification. J. Virol. Methods 2014, 205, 24–30. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, I.S.; Ju, H.J.; Jeong, R.D. Rapid and visual detection of tomato spotted wilt virus using recombinase polymerase amplification combined with lateral flow strips. Mol. Cell. Probes 2021, 57, 101727. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Safenkova, I.V.; Zherdev, A.V.; Dzantiev, B.B. Multiplex Assay of Viruses Integrating Recombinase Polymerase Amplification, Barcode-Anti-Barcode Pairs, Blocking Anti-Primers, and Lateral Flow Assay. Anal. Chem. 2021, 93, 13641–13650. [Google Scholar] [CrossRef]
- Marques, M.C.; Sanchez-Vicente, J.; Ruiz, R.; Montagud-Martinez, R.; Marquez-Costa, R.; Gomez, G.; Carbonell, A.; Daros, J.A.; Rodrigo, G. Diagnostics of Infections Produced by the Plant Viruses TMV, TEV, and PVX with CRISPR-Cas12 and CRISPR-Cas13. ACS Synth. Biol. 2022, 11, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Ma, W.; Jiao, Z.; Tian, Y.; Ismail, R.G.; Zhou, T.; Fan, Z. Reverse transcription-recombinase-aided amplification and CRISPR/Cas12a-based visual detection of maize chlorotic mottle virus. Phytopathol. Res. 2022, 4, 23. [Google Scholar] [CrossRef]
- Lei, R.; Kuang, R.; Peng, X.; Jiao, Z.; Zhao, Z.; Cong, H.; Fan, Z.; Zhang, Y. Portable rapid detection of maize chlorotic mottle virus using RT-RAA/CRISPR-Cas12a based lateral flow assay. Front. Plant Sci. 2023, 14, 1088544. [Google Scholar] [CrossRef]
- Wang, J.; Huang, X.; Chen, S.; Chen, J.; Liang, Z.; Chen, B.; Yang, X.; Zhou, G.; Zhang, T. On-site and visual detection of sorghum mosaic virus and rice stripe mosaic virus based on reverse transcription-recombinase-aided amplification and CRISPR/Cas12a. Front. Genome Ed. 2023, 5, 1124794. [Google Scholar] [CrossRef]
- Cao, Y.; Weng, H.; Rao, S.; Li, J.; Yan, F.; Song, X. Rapid and visual field diagnosis of tomato brown rugose fruit virus using reverse transcription recombinase aided amplification (RT RAA) combined with lateral flow strips (LFS). Crop Prot. 2023, 173, 106355. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, S.; Dong, Z.; Fan, Q.; Lei, R.; Kuang, R.; Zhang, Y. One-Step Reverse-Transcription Recombinase-Aided Amplification CRISPR/Cas12a-Based Lateral Flow Assay for Fast Field Screening and Accurate Differentiation of Four Major Tobamoviruses Infecting Tomato and Pepper. J. Agric. Food Chem. 2023, 71, 10725–10735. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Kokane, S.B.; Kokane, A.D.; Warghane, A.J.; Motghare, M.R.; Bhose, S.; Sharma, A.K.; Reddy, M.K. Development of a recombinase polymerase based isothermal amplification combined with lateral flow assay (HLB-RPA-LFA) for rapid detection of Candidatus Liberibacter asiaticus. PLoS ONE 2018, 13, e0208530. [Google Scholar] [CrossRef] [PubMed]
- Redinbaugh, M.G.; Stewart, L.R. Maize Lethal Necrosis: An Emerging, Synergistic Viral Disease. Annu. Rev. Virol. 2018, 5, 301–322. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Frey, T.S.; Barriball, K.; Paul, P.A.; Willie, K.; Mezzalama, M.; Kimani, E.; Mugambi, C.; Wangai, A.; Prasanna, B.M.; et al. Detection of Diverse Maize Chlorotic Mottle Virus Isolates in Maize Seed. Plant Dis. 2021, 105, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, C.; Liu, F.; Wang, Y.; Li, R.; Chen, G. Recombinase-aided amplification combined with lateral flow dipstick for the rapid detection of Amphidinium carterae. J. Appl. Phycol. 2022, 34, 435–447. [Google Scholar] [CrossRef]
- Wong, N.C.K.; Meshkinfamfard, S.; Turbe, V.; Whitaker, M.; Moshe, M.; Bardanzellu, A.; Dai, T.; Pignatelli, E.; Barclay, W.; Darzi, A.; et al. Machine learning to support visual auditing of home-based lateral flow immunoassay self-test results for SARS-CoV-2 antibodies. Commun. Med. 2022, 2, 78. [Google Scholar] [CrossRef]
- Tong, H.; Cao, C.; You, M.; Han, S.; Liu, Z.; Xiao, Y.; He, W.; Liu, C.; Peng, P.; Xue, Z.; et al. Artificial intelligence-assisted colorimetric lateral flow immunoassay for sensitive and quantitative detection of COVID-19 neutralizing antibody. Biosens. Bioelectron. 2022, 213, 114449. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Amplification | Virus | Type of Viral Genome | Testing Duration | Sensitivity * | Ref. |
|---|---|---|---|---|---|
| LAMP | Cassava brown streak virus | +ssRNA | 40 min | 2.9 ng total RNA/μL | [12] |
| Ugandan cassava brown streak virus | |||||
| Tobacco rattle virus | +ssRNA | <50 min | 78 pg template/μL RNA | [13] | |
| Chinaberry tree badnavirus 1 | dsRNA | 45 min | 0.5 pg/reaction | [14] | |
| Rice stripe virus | ±RNA | 50 min | 3 copies per reaction | [31] | |
| Rice black-streaked dwarf virus | dsRNA | 50 min | |||
| RPA | Alfalfa mosaic virus | +ssRNA | 30 min | 103 copies of RNA in reaction | [7] |
| Plum pox virus | +ssRNA | 20 min | 1.0 fg transcripts/reaction | [18] | |
| Rice black-streaked dwarf virus | dsRNA | 20 min | Similar to RT-PCR | [19] | |
| Milk vetch dwarf virus | ssDNA | 30 min | 101 copies per reaction | [20] | |
| Cymbidium mosaic virus | +ssRNA | 30 min | - | [21] | |
| Barley yellow dwarf virus | +ssRNA | 20 min | 100 pg/μL | [22] | |
| Bean pod mottle virus | +ssRNA | <90 min | 500 pg/μL | [23] | |
| Tomato yellow leaf curl virus | ssDNA | 30 min | 0.5 pg DNA per reaction | [24] | |
| Piper yellow mottle virus | dsDNA | 30 min | 10 times more sensitive than PCR | [25] | |
| Tomato chlorotic spot virus | ±RNA | 15 min | 6 pg/μL of total RNA | [27] | |
| Actinidia chlorotic ringspot-associated virus | +ssRNA | <40 min | 20 viral copies | [28] | |
| Citrus tristeza virus | +ssRNA | 15–20 min | 141 fg of RNA when cDNA used as a template | [30] | |
| Little cherry virus 2 | +ssRNA | - | Similar to RT-PCR | [32] | |
| Tomato spotted wilt virus | ±RNA | 15 min | 10 fg TSWV CP transcripts | [33] | |
| Potato virus Y (PVY) | +ssRNA | 30 min | 4 ng of PVY per g of plant leaves | [34] | |
| Potato virus S (PVS) | +ssRNA | 30 min | 0.04 ng of PVS per g of plant leaves | [34] | |
| Potato leafroll virus (PLRV) | +ssRNA | 30 min | 0.04 ng of PVS per g of plant leaves | [34] | |
| Tobacco mosaic virus | +ssRNA | 40 min | - | [35] | |
| Tobacco etch virus | +ssRNA | 40 min | - | [35] | |
| Potato virus X | +ssRNA | 40 min | - | [35] | |
| RAA | Maize chlorotic mottle virus | +ssRNA | 45 min | 0.02 ng of total RNA | [36,37] |
| Sorghum mosaic virus | +ssRNA | 30 min | 107 dilution | [38] | |
| Rice stripe mosaic virus | -ssRNA | 30 min | 107 dilution | [38] | |
| Tomato brown rugose fruit virus | +ssRNA | 20 min | 101 copies /reaction | [39] | |
| Pepper mild mottle virus | +ssRNA | <1 h | - | [40] | |
| Tomato mosaic virus | [40] | ||||
| Tomato mottle mosaic virus | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Cao, Y.; Yan, F. Isothermal Nucleic Acid Amplification-Based Lateral Flow Testing for the Detection of Plant Viruses. Int. J. Mol. Sci. 2024, 25, 4237. https://doi.org/10.3390/ijms25084237
Song X, Cao Y, Yan F. Isothermal Nucleic Acid Amplification-Based Lateral Flow Testing for the Detection of Plant Viruses. International Journal of Molecular Sciences. 2024; 25(8):4237. https://doi.org/10.3390/ijms25084237
Chicago/Turabian StyleSong, Xuemei, Yuhao Cao, and Fei Yan. 2024. "Isothermal Nucleic Acid Amplification-Based Lateral Flow Testing for the Detection of Plant Viruses" International Journal of Molecular Sciences 25, no. 8: 4237. https://doi.org/10.3390/ijms25084237
APA StyleSong, X., Cao, Y., & Yan, F. (2024). Isothermal Nucleic Acid Amplification-Based Lateral Flow Testing for the Detection of Plant Viruses. International Journal of Molecular Sciences, 25(8), 4237. https://doi.org/10.3390/ijms25084237