

Exploring Advanced Therapies for Primary Biliary Cholangitis: Insights from the Gut Microbiota–Bile Acid–Immunity Network
 , , , and
, , , and
Abstract
:1. Introduction
2. Methods
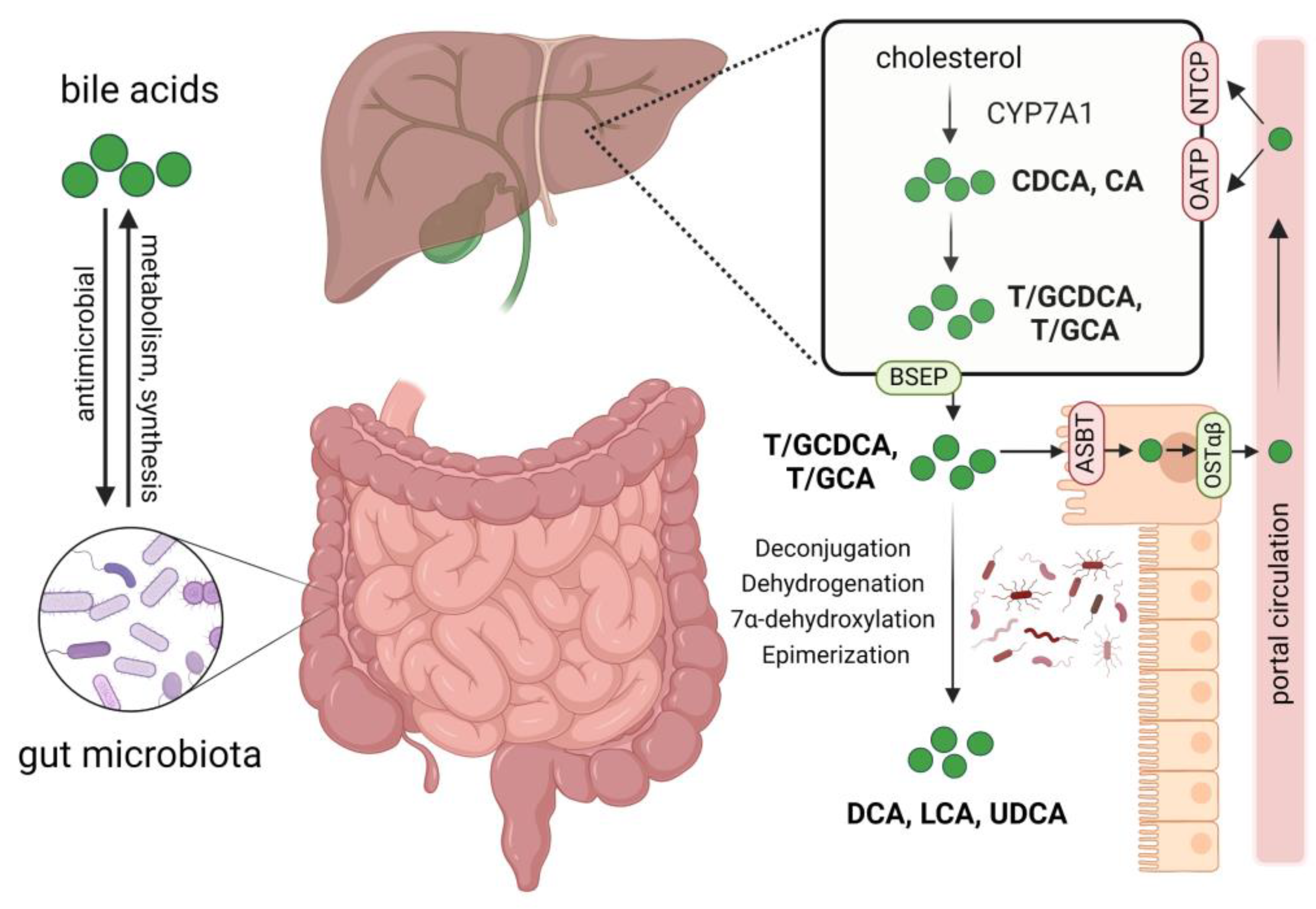
3. Gut Microbiota and Bile Acids in PBC
3.1. Dysbiosis of Gut Microbiota and Impaired Intestinal Barrier in PBC
3.2. Imbalance of Bile Acids in PBC
3.3. Immune Response Mediated by Gut Microbiota and Bile Acids
4. Present and Potential Therapy
4.1. Targeting Gut Microbiota
4.1.1. Antibiotics
4.1.2. Probiotics, Prebiotics, and Synbiotics
4.1.3. Fecal Microbiota Transplantation
4.1.4. Bacteriophages
4.2. Targeting Bile Acids Homeostasis
4.2.1. UDCA
4.2.2. FXR-FGF19 Axis
4.2.3. PPAR Agonists
4.2.4. ASBT Inhibitors
4.3. Targeting Immune Factors Related to Gut Microbiota and Bile Acid
4.3.1. TGR5
4.3.2. Microbial-Derived Molecules
4.3.3. Immune Cells and Signaling
4.4. Targeting the Interaction of Bile Acid and Gut Microbiota
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Trivella, J.; John, B.V.; Levy, C. Primary biliary cholangitis: Epidemiology, prognosis, and treatment. Hepatol. Commun. 2023, 7, e0179. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ma, X.; Efe, C.; Wang, G.; Jeong, S.H.; Abe, K.; Duan, W.; Chen, S.; Kong, Y.; Zhang, D.; et al. APASL clinical practice guidance: The diagnosis and management of patients with primary biliary cholangitis. Hepatol. Int. 2022, 16, 1–23. [Google Scholar] [CrossRef]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef]
- Li, H.; Guan, Y.; Han, C.; Zhang, Y.; Liu, Q.; Wei, W.; Ma, Y. The pathogenesis, models and therapeutic advances of primary biliary cholangitis. Biomed. Pharmacother. 2021, 140, 111754. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Blesl, A.; Stadlbauer, V. The Gut-Liver Axis in Cholestatic Liver Diseases. Nutrients 2021, 13, 1018. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Schnabl, B. The gut-liver axis and gut microbiota in health and liver disease. Nat. Rev. Microbiol. 2023, 21, 719–733. [Google Scholar] [CrossRef]
- Isaacs-Ten, A.; Echeandia, M.; Moreno-Gonzalez, M.; Brion, A.; Goldson, A.; Philo, M.; Patterson, A.M.; Parker, A.; Galduroz, M.; Baker, D.; et al. Intestinal Microbiome-Macrophage Crosstalk Contributes to Cholestatic Liver Disease by Promoting Intestinal Permeability in Mice. Hepatology 2020, 72, 2090–2108. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Shi, Q.; Yuan, X.; Zeng, Y.; Wang, J.; Zhang, Y.; Xue, C.; Li, L. Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease. Nutrients 2023, 15, 2411. [Google Scholar] [CrossRef]
- Lv, L.X.; Fang, D.Q.; Shi, D.; Chen, D.Y.; Yan, R.; Zhu, Y.X.; Chen, Y.F.; Shao, L.; Guo, F.F.; Wu, W.R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive Analysis of Serum and Fecal Bile Acid Profiles and Interaction with Gut Microbiota in Primary Biliary Cholangitis. Clin. Rev. Allergy Immunol. 2020, 58, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Moriya, K.; Nakayama, J.; Inoue, T.; Momoda, R.; Kawaratani, H.; Namisaki, T.; Sato, S.; Douhara, A.; Kaji, K.; et al. Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol. Res. 2020, 50, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Ying, G.X.; Dong, S.L.; Xiang, B.; Jin, Q.F. Gut microbial profile of treatment-naive patients with primary biliary cholangitis. Front. Immunol. 2023, 14, 1126117. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Huang, C.; Zhang, Q.; Tao, S.; Hu, X.; Xu, J.; Jiang, R.; Xu, B.; Liu, Y.; Hou, J. Alterations in gut microbiota and elevated serum bilirubin in primary biliary cholangitis patients treated with ursodeoxycholic acid. Eur. J. Clin. Invest. 2022, 52, e13714. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Shin, A.; Xu, H.; Hemmerich, C.; O’Connell, T.M.; Chalasani, N. Short-chain fatty acid and fecal microbiota profiles are linked to fibrosis in primary biliary cholangitis. FEMS Microbiol. Lett. 2021, 368, fnab038. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Takahashi, A.; Fujita, M.; Imaizumi, H.; Hayashi, M.; Okai, K.; Ohira, H. Dysbiosis of oral microbiota and its association with salivary immunological biomarkers in autoimmune liver disease. PLoS ONE 2018, 13, e0198757. [Google Scholar] [CrossRef] [PubMed]
- Kitahata, S.; Yamamoto, Y.; Yoshida, O.; Tokumoto, Y.; Kawamura, T.; Furukawa, S.; Kumagi, T.; Hirooka, M.; Takeshita, E.; Abe, M.; et al. Ileal mucosa-associated microbiota overgrowth associated with pathogenesis of primary biliary cholangitis. Sci. Rep. 2021, 11, 19705. [Google Scholar] [CrossRef] [PubMed]
- Chascsa, D.M.H.; Lindor, K.D. Emerging therapies for PBC. J. Gastroenterol. 2020, 55, 261–272. [Google Scholar] [CrossRef]
- Mayo, M.J. Mechanisms and molecules: What are the treatment targets for primary biliary cholangitis? Hepatology 2022, 76, 518–531. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef]
- Ma, H.D.; Zhao, Z.B.; Ma, W.T.; Liu, Q.Z.; Gao, C.Y.; Li, L.; Wang, J.; Tsuneyama, K.; Liu, B.; Zhang, W.; et al. Gut microbiota translocation promotes autoimmune cholangitis. J. Autoimmun. 2018, 95, 47–57. [Google Scholar] [CrossRef]
- Schrumpf, E.; Kummen, M.; Valestrand, L.; Greiner, T.U.; Holm, K.; Arulampalam, V.; Reims, H.M.; Baines, J.; Bäckhed, F.; Karlsen, T.H.; et al. The gut microbiota contributes to a mouse model of spontaneous bile duct inflammation. J. Hepatol. 2017, 66, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Jiang, H.; Chen, X.; Wang, Q.; Wang, K.; Ye, J.; Li, Y.; Fang, D.; Lu, Y.; Yang, L.; et al. The Salivary Microbiota of Patients with Primary Biliary Cholangitis Is Distinctive and Pathogenic. Front. Immunol. 2021, 12, 713647. [Google Scholar] [CrossRef]
- Nakamoto, N.; Sasaki, N.; Aoki, R.; Miyamoto, K.; Suda, W.; Teratani, T.; Suzuki, T.; Koda, Y.; Chu, P.S.; Taniki, N.; et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 2019, 4, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Wang, R.; Luo, J.; Ren, F.; Gu, Z.; Zhao, Y.; Zhao, L. The Core and Distinction of the Gut Microbiota in Chinese Populations across Geography and Ethnicity. Microorganisms 2020, 8, 1579. [Google Scholar] [CrossRef]
- Luo, X.; You, X. Genetic predisposition of the gastrointestinal microbiome and primary biliary cholangitis: A bi-directional, two-sample Mendelian randomization analysis. Front. Endocrinol. 2023, 14, 1225742. [Google Scholar] [CrossRef]
- Pabst, O.; Hornef, M.W.; Schaap, F.G.; Cerovic, V.; Clavel, T.; Bruns, T. Gut-liver axis: Barriers and functional circuits. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Meddings, J.; Heathcote, E.J. Abnormal intestinal permeability in primary biliary cirrhosis. Dig. Dis. Sci. 2006, 51, 1607–1613. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, S.; Zhou, G.; Liang, L.; Guo, X.; Mao, P.; Zhou, X.; Wang, H.; Nan, Y.; Xu, D.; et al. Altered biliary epithelial cell and monocyte responses to lipopolysaccharide as a TLR ligand in patients with primary biliary cirrhosis. Scand. J. Gastroenterol. 2011, 46, 485–494. [Google Scholar] [CrossRef]
- Haruta, I.; Hashimoto, E.; Kato, Y.; Kikuchi, K.; Kato, H.; Yagi, J.; Uchiyama, T.; Kobayash, M.; Shiratori, K. Lipoteichoic acid may affect the pathogenesis of bile duct damage in primary biliary cirrhosis. Autoimmunity 2006, 39, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Baragiotta, A.; Pizzuti, D.; Martines, D.; Cecchetto, A.; Chiarelli, S. Mucosal IgA defect in primary biliary cirrhosis. Am. J. Gastroenterol. 2002, 97, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Albers, S.; Trautwein, C. Role of bile acids in the gut-liver axis. J. Hepatol. 2018, 68, 1083–1085. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.F. Role of the anion exchanger 2 in the pathogenesis and treatment of primary biliary cirrhosis. Dig. Dis. 2011, 29, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hisamoto, S.; Shimoda, S.; Harada, K.; Iwasaka, S.; Onohara, S.; Chong, Y.; Nakamura, M.; Bekki, Y.; Yoshizumi, T.; Ikegami, T.; et al. Hydrophobic bile acids suppress expression of AE2 in biliary epithelial cells and induce bile duct inflammation in primary biliary cholangitis. J. Autoimmun. 2016, 75, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gili, L.; Pechlivanis, A.; McDonald, J.A.K.; Begum, S.; Badrock, J.; Dyson, J.K.; Jones, R.; Hirschfield, G.; Ryder, S.D.; Sandford, R.; et al. Bacterial and metabolic phenotypes associated with inadequate response to ursodeoxycholic acid treatment in primary biliary cholangitis. Gut Microbes 2023, 15, 2208501. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Pathogen infections and primary biliary cholangitis. Clin. Exp. Immunol. 2019, 195, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Bowlus, C.L.; Yang, G.X.; Invernizzi, P.; Podda, M.; Van de Water, J.; Ansari, A.A.; Coppel, R.L.; Worman, H.J.; Gores, G.J.; et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 2010, 52, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e437. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. The Role of Butyrate in Attenuating Pathobiont-Induced Hyperinflammation. Immune Netw. 2020, 20, e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Chen, Y.; Wang, Q.; Yan, L.; Wang, R.; Wei, Y.; You, Z.; Li, Y.; Miao, Q.; et al. Alterations in microbiota and their metabolites are associated with beneficial effects of bile acid sequestrant on icteric primary biliary Cholangitis. Gut Microbes 2021, 13, 1946366. [Google Scholar] [CrossRef]
- Gong, Z.; Zhou, J.; Zhao, S.; Tian, C.; Wang, P.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget 2016, 7, 83951–83963. [Google Scholar] [CrossRef]
- Chen, L.; Jiao, T.; Liu, W.; Luo, Y.; Wang, J.; Guo, X.; Tong, X.; Lin, Z.; Sun, C.; Wang, K.; et al. Hepatic cytochrome P450 8B1 and cholic acid potentiate intestinal epithelial injury in colitis by suppressing intestinal stem cell renewal. Cell Stem Cell 2022, 29, 1366–1381.e1369. [Google Scholar] [CrossRef]
- Wu, K.; Liu, Y.; Xia, J.; Liu, J.; Wang, K.; Liang, H.; Xu, F.; Liu, D.; Nie, D.; Tang, X.; et al. Loss of SLC27A5 Activates Hepatic Stellate Cells and Promotes Liver Fibrosis via Unconjugated Cholic Acid. Adv. Sci. 2024, 11, e2304408. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670.e655. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Rahimpour, S.; Nasiri-Toosi, M.; Khalili, H.; Ebrahimi-Daryani, N.; Nouri-Taromlou, M.K.; Azizi, Z. A Triple Blinded, Randomized, Placebo-Controlled Clinical Trial to Evaluate the Efficacy and Safety of Oral Vancomycin in Primary Sclerosing Cholangitis: A Pilot Study. J. Gastrointestin. Liver Dis. 2016, 25, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.H.; Damman, J.; Shah, S.B.; Davies, Y.; Hurwitz, M.; Stephen, M.; Lemos, L.M.; Carey, E.J.; Lindor, K.D.; Buness, C.W.; et al. Open-label prospective therapeutic clinical trials: Oral vancomycin in children and adults with primary sclerosing cholangitis. Scand. J. Gastroenterol. 2020, 55, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.R.; Mack, C.; Mogul, D.; Perito, E.R.; Valentino, P.L.; Amir, A.Z.; DiGuglielmo, M.; Draijer, L.G.; El-Matary, W.; Furuya, K.N.; et al. Oral Vancomycin, Ursodeoxycholic Acid, or No Therapy for Pediatric Primary Sclerosing Cholangitis: A Matched Analysis. Hepatology 2021, 73, 1061–1073. [Google Scholar] [CrossRef]
- Hegade, V.S.; Pechlivanis, A.; McDonald, J.A.K.; Rees, D.; Corrigan, M.; Hirschfield, G.M.; Taylor-Robinson, S.D.; Holmes, E.; Marchesi, J.R.; Kendrick, S.; et al. Autotaxin, bile acid profile and effect of ileal bile acid transporter inhibition in primary biliary cholangitis patients with pruritus. Liver Int. 2019, 39, 967–975. [Google Scholar] [CrossRef]
- Yu, L.; Liu, Y.; Wang, S.; Zhang, Q.; Zhao, J.; Zhang, H.; Narbad, A.; Tian, F.; Zhai, Q.; Chen, W. Cholestasis: Exploring the triangular relationship of gut microbiota-bile acid-cholestasis and the potential probiotic strategies. Gut Microbes 2023, 15, 2181930. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, K.; Li, F.; Gu, Z.; Liu, Q.; He, L.; Shao, T.; Song, Q.; Zhu, F.; Zhang, L.; et al. Probiotic Lactobacillus rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice. Hepatology 2020, 71, 2050–2066. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guan, W.; Zhang, N.; Wang, Y.; Tian, Y.; Sun, H.; Li, X.; Wang, Y.; Liu, J. Lactobacillus plantarum Lp2 improved LPS-induced liver injury through the TLR-4/MAPK/NFκB and Nrf2-HO-1/CYP2E1 pathways in mice. Food Nutr. Res. 2022, 66, 5459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Tian, Y.; Wang, Y.; Fan, Y.; Zhang, Y.; Xing, X.; Nan, B.; Ai, Z.; Li, X.; Wang, Y. Ameliorative effect of Lactobacillus plantarum Lp2 against cyclophosphamide-induced liver injury in mice. Food Chem. Toxicol. 2022, 169, 113433. [Google Scholar] [CrossRef] [PubMed]
- Bomhof, M.R.; Parnell, J.A.; Ramay, H.R.; Crotty, P.; Rioux, K.P.; Probert, C.S.; Jayakumar, S.; Raman, M.; Reimer, R.A. Histological improvement of non-alcoholic steatohepatitis with a prebiotic: A pilot clinical trial. Eur. J. Nutr. 2019, 58, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Behrouz, V.; Aryaeian, N.; Zahedi, M.J.; Jazayeri, S. Effects of probiotic and prebiotic supplementation on metabolic parameters, liver aminotransferases, and systemic inflammation in nonalcoholic fatty liver disease: A randomized clinical trial. J. Food Sci. 2020, 85, 3611–3617. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Afolabi, P.R.; Miles, E.A.; Smith, D.E.; Almehmadi, A.; Alshathry, A.; Childs, C.E.; Del Fabbro, S.; Bilson, J.; Moyses, H.E.; et al. Synbiotics Alter Fecal Microbiomes, But Not Liver Fat or Fibrosis, in a Randomized Trial of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1597–1610.e1597. [Google Scholar] [CrossRef]
- Guery, B.; Galperine, T.; Barbut, F. Clostridioides difficile: Diagnosis and treatments. BMJ 2019, 366, l4609. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Mullish, B.H.; Kelly, C.; Fischer, M. The evolution of the use of faecal microbiota transplantation and emerging therapeutic indications. Lancet 2019, 394, 420–431. [Google Scholar] [CrossRef]
- Zhao, Y.; Gong, C.; Xu, J.; Chen, D.; Yang, B.; Chen, Z.; Wei, L. Research Progress of Fecal Microbiota Transplantation in Liver Diseases. J. Clin. Med. 2023, 12, 1683. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients With Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Nakamoto, N.; Kredo-Russo, S.; Weinstock, E.; Weiner, I.N.; Khabra, E.; Ben-Ishai, N.; Inbar, D.; Kowalsman, N.; Mordoch, R.; et al. Bacteriophage therapy against pathological Klebsiella pneumoniae ameliorates the course of primary sclerosing cholangitis. Nat. Commun. 2023, 14, 3261. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K.V.; Milkiewicz, P.; Janczewska, E.; Malova, E.S.; Sanni, J.; et al. Farnesoid X receptor agonist tropifexor attenuates cholestasis in a randomised trial in patients with primary biliary cholangitis. JHEP Rep. 2022, 4, 100544. [Google Scholar] [CrossRef] [PubMed]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J.; Weltman, M.; Carey, E.J.; Muir, A.J.; Ling, L.; Rossi, S.J.; et al. NGM282 for Treatment of Patients With Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol. Commun. 2018, 2, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hirohara, J.; Nakano, T.; Matsumoto, K.; Chazouillères, O.; Takikawa, H.; Hansen, B.E.; Carrat, F.; Corpechot, C. Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 2021, 75, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M.; Namisaki, T.; Hashimoto, N.; Kawata, K.; Takahashi, A.; et al. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients With Primary Biliary Cholangitis. Hepatology 2019, 70, 2035–2046. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, G.; Zheng, L.; Sun, R.; Wang, X.; Deng, J.; Jia, G.; Yang, C.; Cui, L.; Guo, C.; et al. Effectiveness of Fenofibrate in Treatment-Naive Patients With Primary Biliary Cholangitis: A Randomized Clinical Trial. Am. J. Gastroenterol. 2023, 118, 1973–1979. [Google Scholar] [CrossRef]
- Wang, L.; Sun, K.; Tian, A.; Liu, Y.; Zhang, M.; Zhou, X.; Han, Y. Fenofibrate improves GLOBE and UK-PBC scores and histological features in primary biliary cholangitis. Minerva Med. 2022, 113, 974–982. [Google Scholar] [CrossRef]
- Li, C.; Zheng, K.; Chen, Y.; He, C.; Liu, S.; Yang, Y.; Li, M.; Zeng, X.; Wang, L.; Zhang, F. A randomized, controlled trial on fenofibrate in primary biliary cholangitis patients with incomplete response to ursodeoxycholic acid. Ther. Adv. Chronic. Dis. 2022, 13, 20406223221114198. [Google Scholar] [CrossRef]
- Mayo, M.J.; Vierling, J.M.; Bowlus, C.L.; Levy, C.; Hirschfield, G.M.; Neff, G.W.; Galambos, M.R.; Gordon, S.C.; Borg, B.B.; Harrison, S.A.; et al. Open-label, clinical trial extension: Two-year safety and efficacy results of seladelpar in patients with primary biliary cholangitis. Aliment. Pharmacol. Ther. 2023, 59, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Bowlus, C.L.; Galambos, M.R.; Aspinall, R.J.; Hirschfield, G.M.; Jones, D.E.J.; Dörffel, Y.; Gordon, S.C.; Harrison, S.A.; Kremer, A.E.; Mayo, M.J.; et al. A phase II, randomized, open-label, 52-week study of seladelpar in patients with primary biliary cholangitis. J. Hepatol. 2022, 77, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Shiffman, M.L.; Gulamhusein, A.; Kowdley, K.V.; Vierling, J.M.; Levy, C.; Kremer, A.E.; Zigmond, E.; Andreone, P.; Gordon, S.C.; et al. Seladelpar efficacy and safety at 3 months in patients with primary biliary cholangitis: ENHANCE, a phase 3, randomized, placebo-controlled study. Hepatology 2023, 78, 397–415. [Google Scholar] [CrossRef]
- Kremer, A.E.; Mayo, M.J.; Hirschfield, G.; Levy, C.; Bowlus, C.L.; Jones, D.E.; Steinberg, A.; McWherter, C.A.; Choi, Y.J. Seladelpar improved measures of pruritus, sleep, and fatigue and decreased serum bile acids in patients with primary biliary cholangitis. Liver Int. 2022, 42, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Bowlus, C.L.; Levy, C.; Akarca, U.S.; Alvares-da-Silva, M.R.; Andreone, P.; Arrese, M.; Corpechot, C.; Francque, S.M.; Heneghan, M.A.; et al. Efficacy and Safety of Elafibranor in Primary Biliary Cholangitis. N. Engl. J. Med. 2023, 390, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Vuppalanchi, R.; Caldwell, S.H.; Pyrsopoulos, N.; deLemos, A.S.; Rossi, S.; Levy, C.; Goldberg, D.S.; Mena, E.A.; Sheikh, A.; Ravinuthala, R.; et al. Proof-of-concept study to evaluate the safety and efficacy of saroglitazar in patients with primary biliary cholangitis. J. Hepatol. 2022, 76, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Kendrick, S.; Bowlus, C.L.; Tanaka, A.; Jones, D.; Kremer, A.E.; Mayo, M.J.; Haque, N.; von Maltzahn, R.; Allinder, M.; et al. GLIMMER: A Randomized Phase 2b Dose-Ranging Trial of Linerixibat in Primary Biliary Cholangitis Patients With Pruritus. Clin. Gastroenterol Hepatol. 2023, 21, 1902–1912.e1913. [Google Scholar] [CrossRef] [PubMed]
- Hegade, V.S.; Kendrick, S.F.; Dobbins, R.L.; Miller, S.R.; Thompson, D.; Richards, D.; Storey, J.; Dukes, G.E.; Corrigan, M.; Oude Elferink, R.P.; et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: A double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet 2017, 389, 1114–1123. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef]
- EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [CrossRef]
- Harms, M.H.; van Buuren, H.R.; Corpechot, C.; Thorburn, D.; Janssen, H.L.A.; Lindor, K.D.; Hirschfield, G.M.; Parés, A.; Floreani, A.; Mayo, M.J.; et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 2019, 71, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Dilger, K.; Hohenester, S.; Winkler-Budenhofer, U.; Bastiaansen, B.A.; Schaap, F.G.; Rust, C.; Beuers, U. Effect of ursodeoxycholic acid on bile acid profiles and intestinal detoxification machinery in primary biliary cirrhosis and health. J. Hepatol. 2012, 57, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, S.; Kawata, K.; Ohta, K.; Chida, T.; Suzuki, T.; Tsuneyama, K.; Shimoda, S.; Kurono, N.; Leung, P.S.C.; Gershwin, M.E.; et al. Ursodeoxycholic acid impairs liver-infiltrating T-cell chemotaxis through IFN-γ and CX3CL1 production in primary biliary cholangitis. Eur. J. Immunol. 2021, 51, 1519–1530. [Google Scholar] [CrossRef]
- Paumgartner, G.; Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology 2002, 36, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Xiang, D.; Yang, J.; Liu, L.; Yu, H.; Gong, X.; Liu, D. The regulation of tissue-specific farnesoid X receptor on genes and diseases involved in bile acid homeostasis. Biomed. Pharmacother. 2023, 168, 115606. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol 2015, 185, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Floreani, A.; Gabbia, D.; De Martin, S. Obeticholic Acid for Primary Biliary Cholangitis. Biomedicines 2022, 10, 2464. [Google Scholar] [CrossRef]
- Kjærgaard, K.; Frisch, K.; Sørensen, M.; Munk, O.L.; Hofmann, A.F.; Horsager, J.; Schacht, A.C.; Erickson, M.; Shapiro, D.; Keiding, S. Obeticholic acid improves hepatic bile acid excretion in patients with primary biliary cholangitis. J. Hepatol. 2021, 74, 58–65. [Google Scholar] [CrossRef]
- Friedman, E.S.; Li, Y.; Shen, T.D.; Jiang, J.; Chau, L.; Adorini, L.; Babakhani, F.; Edwards, J.; Shapiro, D.; Zhao, C.; et al. FXR-Dependent Modulation of the Human Small Intestinal Microbiome by the Bile Acid Derivative Obeticholic Acid. Gastroenterology 2018, 155, 1741–1752.e1745. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Parés, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. Fibrates and cholestasis. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef]
- Wang, C.; Shi, Y.; Wang, X.; Ma, H.; Liu, Q.; Gao, Y.; Niu, J. Peroxisome Proliferator-Activated Receptors Regulate Hepatic Immunity and Assist in the Treatment of Primary Biliary Cholangitis. Front. Immunol. 2022, 13, 940688. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Bowlus, C.L.; Mayo, M.J.; Kremer, A.E.; Vierling, J.M.; Kowdley, K.V.; Levy, C.; Villamil, A.; Ladrón de Guevara Cetina, A.L.; Janczewska, E.; et al. A Phase 3 Trial of Seladelpar in Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 783–794. [Google Scholar] [CrossRef]
- Hruz, P.; Zimmermann, C.; Gutmann, H.; Degen, L.; Beuers, U.; Terracciano, L.; Drewe, J.; Beglinger, C. Adaptive regulation of the ileal apical sodium dependent bile acid transporter (ASBT) in patients with obstructive cholestasis. Gut 2006, 55, 395–402. [Google Scholar] [CrossRef]
- Baghdasaryan, A.; Fuchs, C.D.; Österreicher, C.H.; Lemberger, U.J.; Halilbasic, E.; Påhlman, I.; Graffner, H.; Krones, E.; Fickert, P.; Wahlström, A.; et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J. Hepatol. 2016, 64, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Trauner, M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat Rev Gastroenterol. Hepatol. 2022, 19, 432–450. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Fiorucci, C.; Marchianò, S.; Di Giorgio, C.; Roselli, R.; Magro, M.; Distrutti, E.; Bereshchenko, O.; Scarpelli, P.; et al. GPBAR1 Functions as Gatekeeper for Liver NKT Cells and provides Counterregulatory Signals in Mouse Models of Immune-Mediated Hepatitis. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 447–473. [Google Scholar] [CrossRef]
- Duan, S.; Li, X.; Fan, G.; Liu, R. Targeting bile acid signaling for the treatment of liver diseases: From bench to bed. Biomed. Pharmacother. 2022, 152, 113154. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Marinelli, L.; Blottière, H.M.; Larraufie, P.; Lapaque, N. SCFA: Mechanisms and functional importance in the gut. Proc. Nutr. Soc. 2021, 80, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Jena, P.K.; Hu, Y.; Liu, H.X.; Nagar, N.; Kalanetra, K.M.; French, S.W.; French, S.W.; Mills, D.A.; Wan, Y.Y. Hepatic inflammation caused by dysregulated bile acid synthesis is reversible by butyrate supplementation. J. Pathol. 2017, 243, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, J.; Hao, W.; Zhu, H.; Liang, N.; He, Z.; Ma, K.Y.; Chen, Z.Y. Structure-Specific Effects of Short-Chain Fatty Acids on Plasma Cholesterol Concentration in Male Syrian Hamsters. J. Agric. Food Chem. 2017, 65, 10984–10992. [Google Scholar] [CrossRef]
- Shah, S.; Fillier, T.; Pham, T.H.; Thomas, R.; Cheema, S.K. Intraperitoneal Administration of Short-Chain Fatty Acids Improves Lipid Metabolism of Long-Evans Rats in a Sex-Specific Manner. Nutrients 2021, 13, 892. [Google Scholar] [CrossRef]
- Fu, H.Y.; Xu, J.M.; Ai, X.; Dang, F.T.; Tan, X.; Yu, H.Y.; Feng, J.; Yang, W.X.; Ma, H.T.; Tu, R.F.; et al. The Clostridium Metabolite P-Cresol Sulfate Relieves Inflammation of Primary Biliary Cholangitis by Regulating Kupffer Cells. Cells 2022, 11, 3782. [Google Scholar] [CrossRef]
- Glaser, F.; John, C.; Engel, B.; Höh, B.; Weidemann, S.; Dieckhoff, J.; Stein, S.; Becker, N.; Casar, C.; Schuran, F.A.; et al. Liver infiltrating T cells regulate bile acid metabolism in experimental cholangitis. J. Hepatol. 2019, 71, 783–792. [Google Scholar] [CrossRef]
- Huang, B.; Lyu, Z.; Qian, Q.; Chen, Y.; Zhang, J.; Li, B.; Li, Y.; Liang, J.; Liu, Q.; Li, Y.; et al. NUDT1 promotes the accumulation and longevity of CD103(+) T(RM) cells in primary biliary cholangitis. J. Hepatol. 2022, 77, 1311–1324. [Google Scholar] [CrossRef]
- Thompson, M.D.; Moghe, A.; Cornuet, P.; Marino, R.; Tian, J.; Wang, P.; Ma, X.; Abrams, M.; Locker, J.; Monga, S.P.; et al. β-Catenin regulation of farnesoid X receptor signaling and bile acid metabolism during murine cholestasis. Hepatology 2018, 67, 955–971. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Ogawa, E.; Harada, K.; Imamura, J.; Saio, M.; Ikura, Y.; Yatsuhashi, H.; Murata, K.; Miura, K.; Ieiri, I.; et al. Feasibility, safety and tolerability of the CREB-binding protein/β-catenin inhibitor OP-724 in patients with advanced primary biliary cholangitis: An investigator-initiated, open-label, non-randomised, two-centre, phase 1 study. BMJ Open Gastroenterol. 2022, 9, e001001. [Google Scholar] [CrossRef] [PubMed]
- Boicean, A.; Birlutiu, V.; Ichim, C.; Brusnic, O.; Onișor, D.M. Fecal Microbiota Transplantation in Liver Cirrhosis. Biomedicines 2023, 11, 2930. [Google Scholar] [CrossRef]
- Boicean, A.; Birsan, S.; Ichim, C.; Boeras, I.; Roman-Filip, I.; Blanca, G.; Bacila, C.; Fleaca, R.S.; Dura, H.; Roman-Filip, C. Has-miR-129-5p’s Involvement in Different Disorders, from Digestive Cancer to Neurodegenerative Diseases. Biomedicines 2023, 11, 2058. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xing, X.; Xiang, X.; Fan, X.; Men, R.; Ye, T.; Yang, L. MicroRNAs in autoimmune liver diseases: From diagnosis to potential therapeutic targets. Biomed. Pharmacother. 2020, 130, 110558. [Google Scholar] [CrossRef]
- Kim, Y.C.; Jung, H.; Seok, S.; Zhang, Y.; Ma, J.; Li, T.; Kemper, B.; Kemper, J.K. MicroRNA-210 Promotes Bile Acid-Induced Cholestatic Liver Injury by Targeting Mixed-Lineage Leukemia-4 Methyltransferase in Mice. Hepatology 2020, 71, 2118–2134. [Google Scholar] [CrossRef]


{kind=link}
| Studies (Published Time) | Country | Participant (Number) | Sample | Method | Main Findings |
|---|---|---|---|---|---|
| Lv et al. (2016) [11] | China | PBC (42) vs. HC (30) | stool | 16S rRNA | PBC vs. HC: γ-Proteobacteria↑, Enterobacteriaceae↑, Neisseriaceae↑, Spirochaetaceae↑, Veillonella↑, Streptococcus↑, Klebsiella↑, Actinobacillus pleuropneumoniae↑, Anaeroglobus geminatus↑, Enterobacter asburiae↑, Haemophilus parainfluenzae↑, Megasphaera micronuciformis↑, Paraprevotella clara↑; Acidobacteria↓, Lachnobacterium sp. ↓, Bacteroides eggerthii↓, Ruminococcus bromii↓; potential biomarkers: Streptococcus sp. and Veillonella sp. |
| Chen et al. (2020) [12] | China | PBC (79) vs. HC (114) | stool | 16S rRNA | PBC vs. HC: f-Enterobacteriaceae↑, Prevotella↑, Veillonella↑, Fusobacterium↑, Haemophilus↑, Prevotella↑, Streptococcus↑, f-Clostridiaceae↑, Pseudomonas↑, Citrobacter↑, Lactobacillus↑, Salmonella↑, Clostridium↑, Klebsiella↑, Sneathia↑; f-Mogibacteriaceae↓, Blautia↓, f-Christensenellaceae↓, Butyricimonas↓, Akkermansia↓, Odoribacter↓, Dialister↓, f-Rikenellaceae↓, Oscillospira↓, f-S24-7↓, Faecalibacterium↓, Phascolarctobacterium↓, o-Clostridiales↓, Sutterella↓, f-Barnesiellaceae↓, Bacteroides↓ |
| Furukawa et al. (2020) [13] | Japan | PBC (76) vs. HC (23); UDCA non-responder (30) vs. responder (43) | stool | 16S rRNA | PBC vs. HC: diversity↓; Streptococcus↑, Lactobacillus↑, Bifidobacterium↑, Enterococcus↑; Lachnospiraceae↓, Ruminococcaceae↓, Clostridia↓; UDCA non-responder vs. responder: Faecalibacterium↓ |
| Tang et al. (2018) [9] | China | PBC (60) vs. HC (80); PBC before and after UDCA treatment (37) | stool | 16S rRNA | PBC vs. HC: diversity↓ Haemophilus↑, Veillonella↑, Clostridium↑, Lactobacillus↑, Streptococcus↑, Pseudomonas↑, Klebsiella↑, an unknown genus in the family of Enterobacteriaceae↑; Bacteroidetes spp.↓, Sutterella↓, Oscillospira↓, Faecalibacterium↓; PBC after UDCA vs. before UDCA: Haemophilus spp.↓, Streptococcus spp.↓, Pseudomonas spp.↓; Bacteroidetes spp.↑, Sutterella spp.↑, Oscillospira spp.↑ |
| Zhou et al. (2023) [14] | China | PBC (25) vs. HC (25) | stool | 16S rRNA | PBC vs. HC: diversity↓; Acidimicrobiia↑, Yersiniaceae↑, Serratia↑, ucg_010↑; Faecalibacterium↓, Ruminococcaceae↓, Sutterellaceae↓, Oscillospiraceae↓, Parasutterella↓, Clostridia↓, Coprococcus↓, Christensenellaceae↓; PBC with anti-gp210-positive vs. anti-gp210-negative: Oscillospiraceae↓ |
| Han et al. (2022) [15] | China | PBC TB+ (20) vs. TB− * (27) | stool | 16S rRNA | PBC TB+ vs. TB−: diversity↓; Proteobacteria↑; Firmicutes↓, Bacteroidetes↓, Actinobacteria↓, Saccharibacteria↓, Gemmiger↓, Blautia↓, Anaerostipes↓, Coprococcus↓, Holdemania× |
| Lammert et al. (2021) [16] | USA | PBC with non-advanced fibrosis (15) vs. advanced fibrosis ** (8) | stool | 16S rRNA | PBC with advanced fibrosis vs. non-advanced fibrosis: diversity↓; Weissella↑ |
| Abe et al. (2018) [17] | Japan | PBC (39) vs. HC (17) | stool; saliva | 16S rDNA | PBC vs. HC (stool): Lactobacillales↑; Clostridium subcluster XIVa↓; PBC vs. HC (saliva): Veillonella↑, Eubacterium↑; Fusobacterium↓ |
| Kitahata et al. (2021) [18] | Japan | PBC (34) vs. HC (21) | ileal mucosa | 16S rRNA | PBC vs. HC: diversity↓; Sphingomonas↑, Pseudomonas↑, Methylobacterium↑, Carnobacterium↑, Acinetobacter↑, Curvibacter↑, an unknown genus Clostridiaceae↑; Leptotrichia↓, Morganella↓, Lautropia↓, Mogibacterium↓, Atopobium↓, Bulleidia↓, Eikenella↓, Paludibacter↓, an unknown genus belonging to the class TM7_3↓, and an unknown genus of the family F16↓ |
| Agents | Mechanism | Clinical Status/Outcomes |
|---|---|---|
| Approved treatments | ||
| UDCA | Alter BA pool | First-line therapeutic drug |
| OCA | FXR agonist | Second-line therapeutic drug |
| Potential treatments | ||
| Tropifexor (LJN452) | FXR agonist | Reduced GGT and ALP levels, the most frequent adverse event was pruritus [73]; |
| Cilofexor | FXR agonist | Phase 2 (NCT02943447) |
| EDP-305 | FXR agonist | Phase 2 (NCT03394924) |
| Aldafermin (NGM282) | FGF19 agonist | Decreased ALP, GGT, and serum transaminase levels, along with a reduction of C4 and total bile acid levels [74] |
| Bezafibrate | Pan-PPAR agonist | Combination therapy (with UDCA) improved liver biochemistries, GLOBE and UK-PBC scores and long-term prognosis [75,76,77]; Phase 3 (NCT04751188) Phase 2 (NCT04594694) Phase 2 (NCT05239468) |
| Fenofibrate | PPARα agonist | Combination therapy (with UDCA) reduced serum ALP levels; improved GLOBE and UK-PBC scores [78,79,80]; Phase 3 (NCT05751967) Phase 2/3 (NCT05749822) |
| Seladelpar | PPARδ agonist | Improved liver biochemistry and pruritus, decreased C4 and serum bile acids concentration, appeared safe and well tolerated [81,82,83,84]; Phase 3 (NCT06051617) Phase 3 (NCT06060665) Newly approved by FDA |
| Elafibranor | PPARα + PPARδ agonist | Greater improvements in relevant biochemical indicators of cholestasis, such as ALP and total bilirubin levels [85]; Phase 2 (NCT05627362) Phase 3 (NCT06016842) |
| Saroglitazar | PPARα + PPARγ agonist | Rapid and sustained improvements in ALP, but a higher incidence of elevated liver enzymes was observed with the 4 mg dose [86]; Phase 2b/3 (NCT05133336) |
| Linerixibat (GSK2330672) | ASBT inhibitor | Improved pruritus and decreased serum bile acids concentration, the most common adverse event was diarrhea [87,88]; Phase 3 (NCT04950127) |
| Volixibat | ASBT inhibitor | Phase 2 (NCT05050136) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; He, K.; Pang, K.; Yang, D.; Lyu, C.; Xu, H.; Wu, D. Exploring Advanced Therapies for Primary Biliary Cholangitis: Insights from the Gut Microbiota–Bile Acid–Immunity Network. Int. J. Mol. Sci. 2024, 25, 4321. https://doi.org/10.3390/ijms25084321
Guo Z, He K, Pang K, Yang D, Lyu C, Xu H, Wu D. Exploring Advanced Therapies for Primary Biliary Cholangitis: Insights from the Gut Microbiota–Bile Acid–Immunity Network. International Journal of Molecular Sciences. 2024; 25(8):4321. https://doi.org/10.3390/ijms25084321
Chicago/Turabian StyleGuo, Ziqi, Kun He, Ke Pang, Daiyu Yang, Chengzhen Lyu, Haifeng Xu, and Dong Wu. 2024. "Exploring Advanced Therapies for Primary Biliary Cholangitis: Insights from the Gut Microbiota–Bile Acid–Immunity Network" International Journal of Molecular Sciences 25, no. 8: 4321. https://doi.org/10.3390/ijms25084321
APA StyleGuo, Z., He, K., Pang, K., Yang, D., Lyu, C., Xu, H., & Wu, D. (2024). Exploring Advanced Therapies for Primary Biliary Cholangitis: Insights from the Gut Microbiota–Bile Acid–Immunity Network. International Journal of Molecular Sciences, 25(8), 4321. https://doi.org/10.3390/ijms25084321