


Ischemia–Reperfusion Injury in Kidney Transplantation: Mechanisms and Potential Therapeutic Targets

 ,
,  , ,
, ,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
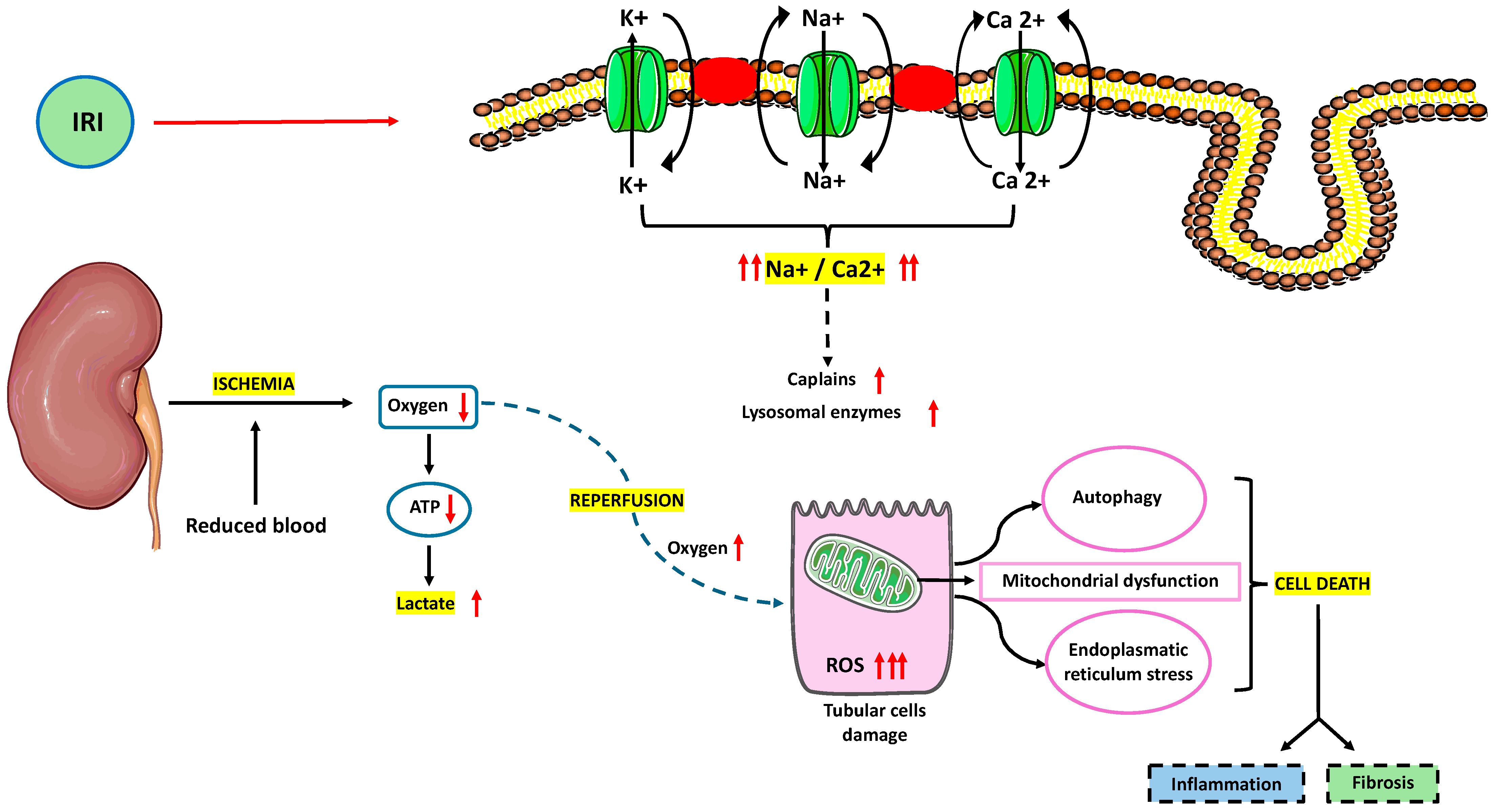
2. Pathophysiology of IRI
3. Metabolic Reprogramming during IRI
4. Immune Response during IRI
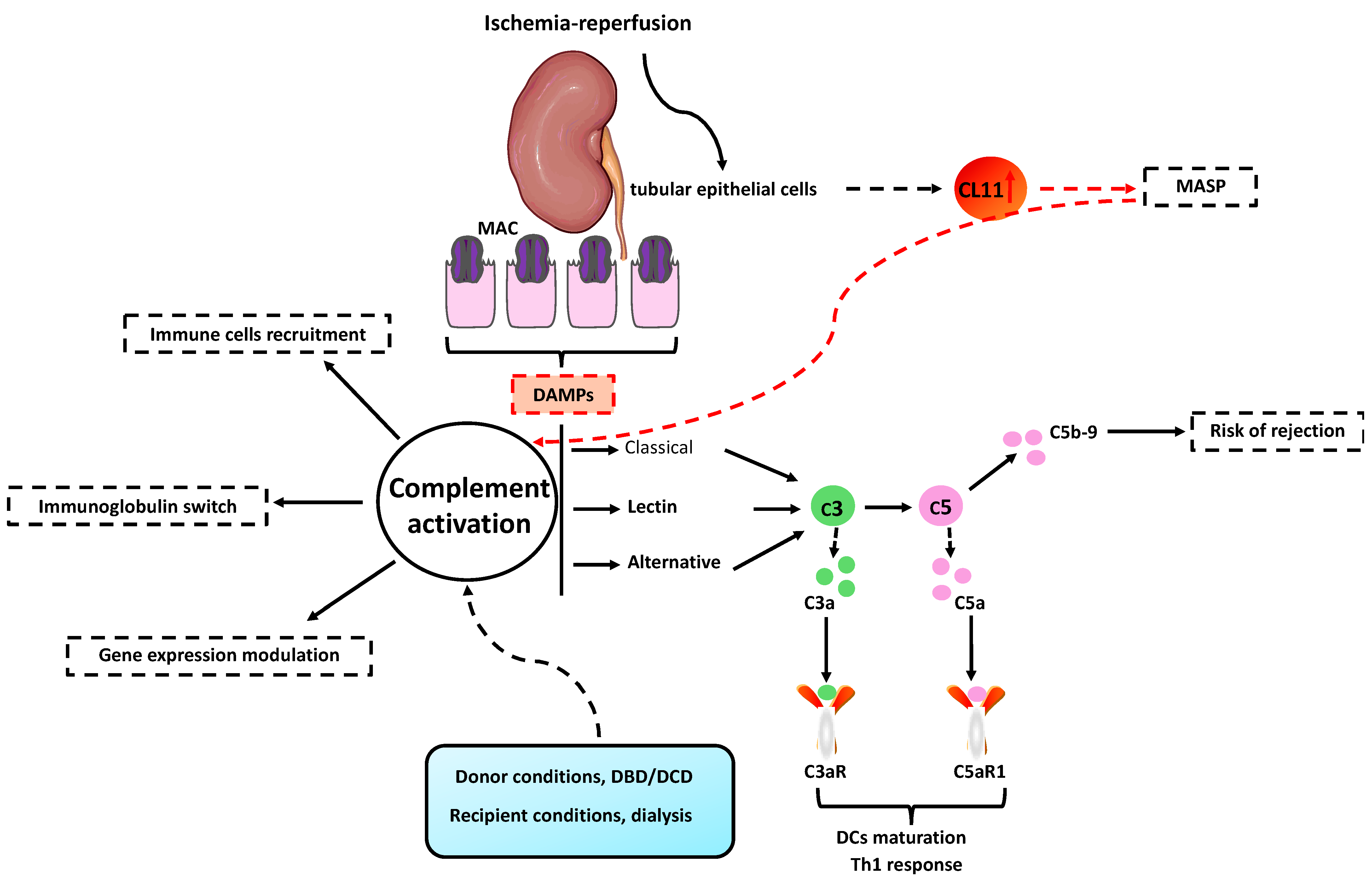
5. Complement System and IRI
6. Prevention and Therapeutic Perspective of IRI
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boenink, R.; Kramer, A.; Tuinhout, R.E.; Savoye, E.; Åsberg, A.; Idrizi, A.; Kerschbaum, J.; Ziedina, I.; Ziginskiene, E.; Farrugia, E.; et al. Trends in Kidney Transplantation Rate across Europe: Study from the ERA Registry. Nephrol. Dial. Transplant. 2023, 38, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Bettocchi, C.; Battaglia, M.; Impedovo, S.V.; Vavallo, A.; Grandaliano, G.; Castellano, G.; Schena, F.P.; Selvaggi, F.P.; Ditonno, P. Extended Criteria Donor Kidney Transplantation: Comparative Outcome Analysis between Single versus Double Kidney Transplantation at 5 Years. Transplant. Proc. 2010, 42, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Vavallo, A.; Lucarelli, G.; Bettocchi, C.; Tedeschi, M.; Palazzo, S.; Losappio, V.; Gesualdo, L.; Grandaliano, G.; Selvaggi, F.P.; Battaglia, M.; et al. Allograft Nephrectomy: What Is the Best Surgical Technique? Transplant. Proc. 2012, 44, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Ditonno, P.; Lucarelli, G.; Impedovo, S.V.; Spilotros, M.; Grandaliano, G.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M. Obesity in Kidney Transplantation Affects Renal Function But Not Graft and Patient Survival. Transplant. Proc. 2011, 43, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Losappio, V.; Stallone, G.; Infante, B.; Schena, A.; Rossini, M.; Maiorano, A.; Fiorentino, M.; Ditonno, P.; Lucarelli, G.; Battaglia, M.; et al. A Single-Center Cohort Study to Define the Role of Pretransplant Biopsy Score in the Long-Term Outcome of Kidney Transplantation. Transplantation 2014, 97, 934–939. [Google Scholar] [CrossRef]
- Loizzo, D.; Di Meo, N.A.; Peluso, M.R.; Rutigliano, M.; Matera, M.; Miacola, C.; Palella, G.; Tedeschi, M.; Spilotros, M.; Ferro, M.; et al. Novel Insights into the Molecular Mechanisms of Ischemia/Reperfusion Injury in Kidney Transplantation. Transplantology 2021, 2, 191–207. [Google Scholar] [CrossRef]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated Necrosis in Kidney Ischemia-Reperfusion Injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef]
- Movahed, M.; Brockie, S.; Hong, J.; Fehlings, M.G. Transcriptomic Hallmarks of Ischemia-Reperfusion Injury. Cells 2021, 10, 1838. [Google Scholar] [CrossRef]
- Park, M.; Kwon, C.H.; Ha, H.K.; Han, M.; Song, S.H. RNA-Seq Identifies Condition-Specific Biological Signatures of Ischemia-Reperfusion Injury in the Human Kidney. BMC Nephrol. 2020, 21, 398. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, C.; Xu, Y.; Hu, X. Comprehensive Molecular and Cellular Characterization of Endoplasmic Reticulum Stress-Related Key Genes in Renal Ischemia/Reperfusion Injury. Front. Immunol. 2024, 15, 1340997. [Google Scholar] [CrossRef]
- Rahbar Saadat, Y.; Hosseiniyan Khatibi, S.M.; Sani, A.; Zununi Vahed, S.; Ardalan, M. Ischemic Tubular Injury: Oxygen-Sensitive Signals and Metabolic Reprogramming. Inflammopharmacology 2023, 31, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Devalaraja-Narashimha, K.; Padanilam, B.J. TIGAR Regulates Glycolysis in Ischemic Kidney Proximal Tubules. Am. J. Physiol.-Ren. Physiol. 2015, 308, F298–F308. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.; Padanilam, B.J.; Jang, H.-S.; Kim, J. 2-Mercaptoethanol Protects against DNA Double-Strand Breaks after Kidney Ischemia and Reperfusion Injury through GPX4 Upregulation. Pharmacol. Rep. 2022, 74, 1041–1053. [Google Scholar] [CrossRef]
- Kim, J. Spermidine Rescues Proximal Tubular Cells from Oxidative Stress and Necrosis after Ischemic Acute Kidney Injury. Arch. Pharm. Res. 2017, 40, 1197–1208. [Google Scholar] [CrossRef]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and Mitophagy in Acute Kidney Injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; Di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef] [PubMed]
- Grossi, V.; Lucarelli, G.; Forte, G.; Peserico, A.; Matrone, A.; Germani, A.; Rutigliano, M.; Stella, A.; Bagnulo, R.; Loconte, D.; et al. Loss of STK11 Expression Is an Early Event in Prostate Carcinogenesis and Predicts Therapeutic Response to Targeted Therapy against MAPK/P38. Autophagy 2015, 11, 2102–2113. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Boya, P. BNIP3L/NIX-Dependent Mitophagy Regulates Cell Differentiation via Metabolic Reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 Is Able to Induce Mitophagy via LC3 Binding, Regardless of PARKIN and P62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-Mediated Mitophagy Protects against Renal Ischemia-Reperfusion Injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 Pathway of Mitophagy Is Activated to Protect against Renal Ischemia-Reperfusion Injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.-M.; Dong, Z. Clearance of Damaged Mitochondria via Mitophagy Is Important to the Protective Effect of Ischemic Preconditioning in Kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.-H.; Guo, M. MUL1 Acts in Parallel to the PINK1/Parkin Pathway in Regulating Mitofusin and Compensates for Loss of PINK1/Parkin. Elife 2014, 3, e01958. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy Receptor FUNDC1 Regulates Mitochondrial Dynamics and Mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhu, J.; Zhang, M.; Luo, Y.; Li, Y.; Shi, L.; Huang, J.; Shadekejiang, H.; Dong, S.; Wu, X. OGG1 Aggravates Renal Ischemia–Reperfusion Injury by Repressing PINK1 -mediated Mitophagy. Cell Prolif. 2023, 56, e13418. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 Mediates Ferroptosis That Contributes to Renal Ischemia/Reperfusion Injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Huang, L.-L.; Liao, X.-H.; Sun, H.; Jiang, X.; Liu, Q.; Zhang, L. Augmenter of Liver Regeneration Protects the Kidney from Ischaemia-Reperfusion Injury in Ferroptosis. J. Cell Mol. Med. 2019, 23, 4153–4164. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, X.; Cui, S.; Wu, Y.; Zhang, Y.; Shen, X.; Xie, C.; Li, J. Dephosphorylation of AMP-Activated Protein Kinase Exacerbates Ischemia/Reperfusion-Induced Acute Kidney Injury via Mitochondrial Dysfunction. Kidney Int. 2022, 101, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Mechanisms Leading to Differential Hypoxia-Inducible Factor Signaling in the Diabetic Kidney: Modulation by SGLT2 Inhibitors and Hypoxia Mimetics. Am. J. Kidney Dis. 2021, 77, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Keogh, B.; McGuirk, P.; Parker, A.E. TLR2 and TLR4 in Ischemia Reperfusion Injury. Mediat. Inflamm. 2010, 2010, 704202. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on Ischemia-Reperfusion Injury in Kidney Transplantation: Pathogenesis and Treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 Activation Mediates Kidney Ischemia/Reperfusion Injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef]
- Chen, J.; John, R.; Richardson, J.A.; Shelton, J.M.; Zhou, X.J.; Wang, Y.; Wu, Q.Q.; Hartono, J.R.; Winterberg, P.D.; Lu, C.Y. Toll-like Receptor 4 Regulates Early Endothelial Activation during Ischemic Acute Kidney Injury. Kidney Int. 2011, 79, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Rabb, H. The Innate Immune Response in Ischemic Acute Kidney Injury. Clin. Immunol. 2009, 130, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.D.; Kirschning, C.J.; Akira, S.; van der Poll, T.; Weening, J.J.; Florquin, S. Renal-Associated TLR2 Mediates Ischemia/Reperfusion Injury in the Kidney. J. Clin. Investig. 2005, 115, 2894–2903. [Google Scholar] [CrossRef]
- Rusai, K.; Sollinger, D.; Baumann, M.; Wagner, B.; Strobl, M.; Schmaderer, C.; Roos, M.; Kirschning, C.; Heemann, U.; Lutz, J. Toll-like Receptors 2 and 4 in Renal Ischemia/Reperfusion Injury. Pediatr. Nephrol. 2010, 25, 853–860. [Google Scholar] [CrossRef]
- Shigeoka, A.A.; Holscher, T.D.; King, A.J.; Hall, F.W.; Kiosses, W.B.; Tobias, P.S.; Mackman, N.; McKay, D.B. TLR2 Is Constitutively Expressed within the Kidney and Participates in Ischemic Renal Injury through Both MyD88-Dependent and -Independent Pathways. J. Immunol. 2007, 178, 6252–6258. [Google Scholar] [CrossRef]
- Hasegawa, S.; Inoue, T.; Nakamura, Y.; Fukaya, D.; Uni, R.; Wu, C.-H.; Fujii, R.; Peerapanyasut, W.; Taguchi, A.; Kohro, T.; et al. Activation of Sympathetic Signaling in Macrophages Blocks Systemic Inflammation and Protects against Renal Ischemia-Reperfusion Injury. JASN 2021, 32, 1599–1615. [Google Scholar] [CrossRef]
- Batal, I.; De Serres, S.A.; Safa, K.; Bijol, V.; Ueno, T.; Onozato, M.L.; Iafrate, A.J.; Herter, J.M.; Lichtman, A.H.; Mayadas, T.N.; et al. Dendritic Cells in Kidney Transplant Biopsy Samples Are Associated with T Cell Infiltration and Poor Allograft Survival. J. Am. Soc. Nephrol. 2015, 26, 3102–3113. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; Ditonno, P.; et al. Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma. JCM 2023, 12, 3888. [Google Scholar] [CrossRef]
- Ysebaert, D.K.; De Greef, K.E.; De Beuf, A.; Van Rompay, A.R.; Vercauteren, S.; Persy, V.P.; De Broe, M.E. T Cells as Mediators in Renal Ischemia/Reperfusion Injury. Kidney Int. 2004, 66, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, E.; Kwon, S.-H.; Yu, M.-Y.; Cha, R.-H.; Lee, H.; Kim, D.K.; Lee, J.P.; Ye, S.-K.; Yoo, J.-Y.; et al. Transcriptional Modulation of the T Helper 17/Interleukin 17 Axis Ameliorates Renal Ischemia-Reperfusion Injury. Nephrol. Dial. Transplant. 2019, 34, 1481–1498. [Google Scholar] [CrossRef]
- Loverre, A.; Divella, C.; Castellano, G.; Tataranni, T.; Zaza, G.; Rossini, M.; Ditonno, P.; Battaglia, M.; Palazzo, S.; Gigante, M.; et al. T Helper 1, 2 and 17 Cell Subsets in Renal Transplant Patients with Delayed Graft Function. Transplant. Int. 2011, 24, 233–242. [Google Scholar] [CrossRef]
- Ferrer, I.R.; Hester, J.; Bushell, A.; Wood, K.J. Induction of Transplantation Tolerance through Regulatory Cells: From Mice to Men. Immunol. Rev. 2014, 258, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, Y.M.; Wang, Y.; Zhang, G.Y.; Zheng, G.; Yi, S.; O’Connell, P.J.; Harris, D.C.H.; Alexander, S.I. Regulatory T Cells in Kidney Disease and Transplantation. Kidney Int. 2016, 90, 502–514. [Google Scholar] [CrossRef]
- Fan, H.; Liu, J.; Sun, J.; Feng, G.; Li, J. Advances in the Study of B Cells in Renal Ischemia-Reperfusion Injury. Front. Immunol. 2023, 14, 1216094. [Google Scholar] [CrossRef]
- Inaba, A.; Tuong, Z.K.; Riding, A.M.; Mathews, R.J.; Martin, J.L.; Saeb-Parsy, K.; Clatworthy, M.R. B Lymphocyte-Derived CCL7 Augments Neutrophil and Monocyte Recruitment, Exacerbating Acute Kidney Injury. J. Immunol. 2020, 205, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Li, K.; Anderson, K.; Farrar, C.A.; Lu, B.; Smith, R.A.G.; Sacks, S.H.; Zhou, W. Local Production and Activation of Complement Up-Regulates the Allostimulatory Function of Dendritic Cells through C3a-C3aR Interaction. Blood 2008, 111, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular Complement Activation Sustains T Cell Homeostasis and Mediates Effector Differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4+ T Cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Simone, S.; Gesualdo, L.; Battaglia, M.; Ditonno, P.; et al. Complement System and the Kidney: Its Role in Renal Diseases, Kidney Transplantation and Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 16515. [Google Scholar] [CrossRef] [PubMed]
- Divella, C.; Stasi, A.; Franzin, R.; Rossini, M.; Pontrelli, P.; Sallustio, F.; Netti, G.S.; Ranieri, E.; Lacitignola, L.; Staffieri, F.; et al. Pentraxin-3-Mediated Complement Activation in a Swine Model of Renal Ischemia/Reperfusion Injury. Aging 2021, 13, 10920–10933. [Google Scholar] [CrossRef]
- Poppelaars, F.; Seelen, M.A. Complement-Mediated Inflammation and Injury in Brain Dead Organ Donors. Mol. Immunol. 2017, 84, 77–83. [Google Scholar] [CrossRef]
- Bartoszek, D.; Mazanowska, O.; Kościelska-Kasprzak, K.; Kamińska, D.; Lepiesza, A.; Chudoba, P.; Myszka, M.; Żabińska, M.; Klinger, M. Functional Activity of the Complement System in Deceased Donors in Relation to Kidney Allograft Outcome. Transplant. Proc. 2018, 50, 1697–1700. [Google Scholar] [CrossRef]
- Danobeitia, J.S.; Zens, T.J.; Chlebeck, P.J.; Zitur, L.J.; Reyes, J.A.; Eerhart, M.J.; Coonen, J.; Capuano, S.; D’Alessandro, A.M.; Torrealba, J.R.; et al. Targeted Donor Complement Blockade after Brain Death Prevents Delayed Graft Function in a Nonhuman Primate Model of Kidney Transplantation. Am. J. Transplant. 2020, 20, 1513–1526. [Google Scholar] [CrossRef]
- Jager, N.M.; van Zanden, J.E.; Subías, M.; Leuvenink, H.G.D.; Daha, M.R.; Rodríguez de Córdoba, S.; Poppelaars, F.; Seelen, M.A. Blocking Complement Factor B Activation Reduces Renal Injury and Inflammation in a Rat Brain Death Model. Front. Immunol. 2019, 10, 2528. [Google Scholar] [CrossRef]
- Santarsiero, D.; Aiello, S. The Complement System in Kidney Transplantation. Cells 2023, 12, 791. [Google Scholar] [CrossRef]
- Casiraghi, F.; Azzollini, N.; Todeschini, M.; Fiori, S.; Cavinato, R.A.; Cassis, P.; Solini, S.; Pezzuto, F.; Mister, M.; Thurman, J.M.; et al. Complement Alternative Pathway Deficiency in Recipients Protects Kidney Allograft From Ischemia/Reperfusion Injury and Alloreactive T Cell Response. Am. J. Transplant. 2017, 17, 2312–2325. [Google Scholar] [CrossRef]
- Wu, Y.; Zwaini, Z.D.; Brunskill, N.J.; Zhang, X.; Wang, H.; Chana, R.; Stover, C.M.; Yang, B. Properdin Deficiency Impairs Phagocytosis and Enhances Injury at Kidney Repair Phase Post Ischemia-Reperfusion. Front. Immunol. 2021, 12, 697760. [Google Scholar] [CrossRef]
- Bongoni, A.K.; Lu, B.; Salvaris, E.J.; Roberts, V.; Fang, D.; McRae, J.L.; Fisicaro, N.; Dwyer, K.M.; Cowan, P.J. Overexpression of Human CD55 and CD59 or Treatment with Human CD55 Protects against Renal Ischemia-Reperfusion Injury in Mice. J. Immunol. 2017, 198, 4837–4845. [Google Scholar] [CrossRef]
- Heeger, P.S.; Lalli, P.N.; Lin, F.; Valujskikh, A.; Liu, J.; Muqim, N.; Xu, Y.; Medof, M.E. Decay-Accelerating Factor Modulates Induction of T Cell Immunity. J. Exp. Med. 2005, 201, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Peng, Q.; Xing, G.; Li, K.; Wang, N.; Farrar, C.A.; Meader, L.; Sacks, S.H.; Zhou, W. Deficiency of C5aR Prolongs Renal Allograft Survival. J. Am. Soc. Nephrol. 2010, 21, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Nauser, C.L.; Farrar, C.A.; Sacks, S.H. Complement Recognition Pathways in Renal Transplantation. J. Am. Soc. Nephrol. 2017, 28, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, F.; Stasi, A.; Curci, C.; Divella, C.; Picerno, A.; Franzin, R.; De Palma, G.; Rutigliano, M.; Lucarelli, G.; Battaglia, M.; et al. Renal Progenitor Cells Revert LPS-induced Endothelial-to-mesenchymal Transition by Secreting CXCL6, SAA4, and BPIFA2 Antiseptic Peptides. FASEB J. 2019, 33, 10753–10766. [Google Scholar] [CrossRef]
- Castellano, G.; Franzin, R.; Stasi, A.; Divella, C.; Sallustio, F.; Pontrelli, P.; Lucarelli, G.; Battaglia, M.; Staffieri, F.; Crovace, A.; et al. Complement Activation During Ischemia/Reperfusion Injury Induces Pericyte-to-Myofibroblast Transdifferentiation Regulating Peritubular Capillary Lumen Reduction Through pERK Signaling. Front. Immunol. 2018, 9, 1002. [Google Scholar] [CrossRef]
- Castellano, G.; Franzin, R.; Sallustio, F.; Stasi, A.; Banelli, B.; Romani, M.; De Palma, G.; Lucarelli, G.; Divella, C.; Battaglia, M.; et al. Complement Component C5a Induces Aberrant Epigenetic Modifications in Renal Tubular Epithelial Cells Accelerating Senescence by Wnt4/Βcatenin Signaling after Ischemia/Reperfusion Injury. Aging 2019, 11, 4382–4406. [Google Scholar] [CrossRef]
- Castellano, G.; Intini, A.; Stasi, A.; Divella, C.; Gigante, M.; Pontrelli, P.; Franzin, R.; Accetturo, M.; Zito, A.; Fiorentino, M.; et al. Complement Modulation of Anti-Aging Factor Klotho in Ischemia/Reperfusion Injury and Delayed Graft Function. Am. J. Transplant. 2016, 16, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, X.; Feng, B.; Sun, H.; Suzuki, M.; Ichim, T.; Kubo, N.; Wong, A.; Min, L.R.; Budohn, M.E.; et al. Gene Silencing of Complement C5a Receptor Using siRNA for Preventing Ischemia/Reperfusion Injury. Am. J. Pathol. 2008, 173, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Vonbrunn, E.; Büttner-Herold, M.; Amann, K.; Daniel, C. Complement Inhibition in Kidney Transplantation: Where Are We Now? BioDrugs 2023, 37, 5–19. [Google Scholar] [CrossRef]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A Phase I/II, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Am. J. Transplant. 2018, 18, 2955–2964. [Google Scholar] [CrossRef]
- Brüggenwirth, I.M.A.; Martins, P.N. RNA Interference Therapeutics in Organ Transplantation: The Dawn of a New Era. Am. J. Transplant. 2020, 20, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Q.; Meng, H.; Duan, H.; Liu, X.; Wu, J.; Gao, F.; Wang, S.; Tan, R.; Yuan, J. Ischemia-Reperfusion Injury: Molecular Mechanisms and Therapeutic Targets. Signal Transduct. Target. Ther. 2024, 9, 12. [Google Scholar] [CrossRef]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Autorino, R.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Cancer Stem Cells in Renal Cell Carcinoma: Origins and Biomarkers. Int. J. Mol. Sci. 2023, 24, 13179. [Google Scholar] [CrossRef]
- Zulpaite, R.; Miknevicius, P.; Leber, B.; Strupas, K.; Stiegler, P.; Schemmer, P. Ex-Vivo Kidney Machine Perfusion: Therapeutic Potential. Front. Med. 2021, 8, 808719. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasorsa, F.; Rutigliano, M.; Milella, M.; d’Amati, A.; Crocetto, F.; Pandolfo, S.D.; Barone, B.; Ferro, M.; Spilotros, M.; Battaglia, M.; et al. Ischemia–Reperfusion Injury in Kidney Transplantation: Mechanisms and Potential Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 4332. https://doi.org/10.3390/ijms25084332
Lasorsa F, Rutigliano M, Milella M, d’Amati A, Crocetto F, Pandolfo SD, Barone B, Ferro M, Spilotros M, Battaglia M, et al. Ischemia–Reperfusion Injury in Kidney Transplantation: Mechanisms and Potential Therapeutic Targets. International Journal of Molecular Sciences. 2024; 25(8):4332. https://doi.org/10.3390/ijms25084332
Chicago/Turabian StyleLasorsa, Francesco, Monica Rutigliano, Martina Milella, Antonio d’Amati, Felice Crocetto, Savio Domenico Pandolfo, Biagio Barone, Matteo Ferro, Marco Spilotros, Michele Battaglia, and et al. 2024. "Ischemia–Reperfusion Injury in Kidney Transplantation: Mechanisms and Potential Therapeutic Targets" International Journal of Molecular Sciences 25, no. 8: 4332. https://doi.org/10.3390/ijms25084332
APA StyleLasorsa, F., Rutigliano, M., Milella, M., d’Amati, A., Crocetto, F., Pandolfo, S. D., Barone, B., Ferro, M., Spilotros, M., Battaglia, M., Ditonno, P., & Lucarelli, G. (2024). Ischemia–Reperfusion Injury in Kidney Transplantation: Mechanisms and Potential Therapeutic Targets. International Journal of Molecular Sciences, 25(8), 4332. https://doi.org/10.3390/ijms25084332