
Missing WD40 Repeats in ATG16L1 Delays Canonical Autophagy and Inhibits Noncanonical Autophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Autophagy Induced by Torin1 and Starvation Dropped Due to the Deletion of WDR of ATG16L1
2.2. Deletion of the WDR Domain of ATG16L1 Impaired Basal Autophagic Degradation
2.3. WDR Deletion Does Not Affect the Lysosome Degradation Function and the Fusion between Autophagosome and Lysosome
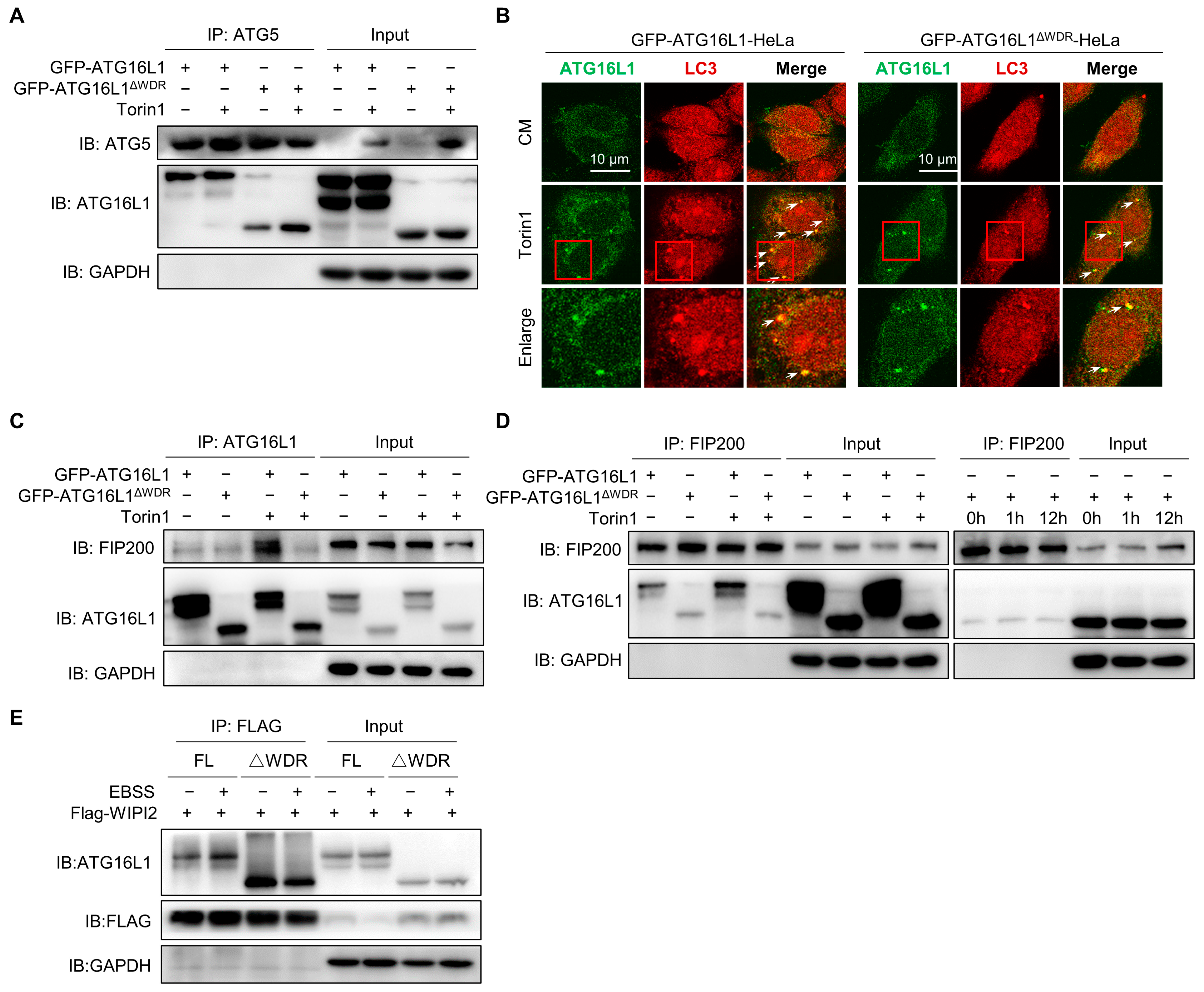
2.4. WDR Deletion Reduces the Interaction of ATG16L1 with FIP200 and WIPI2
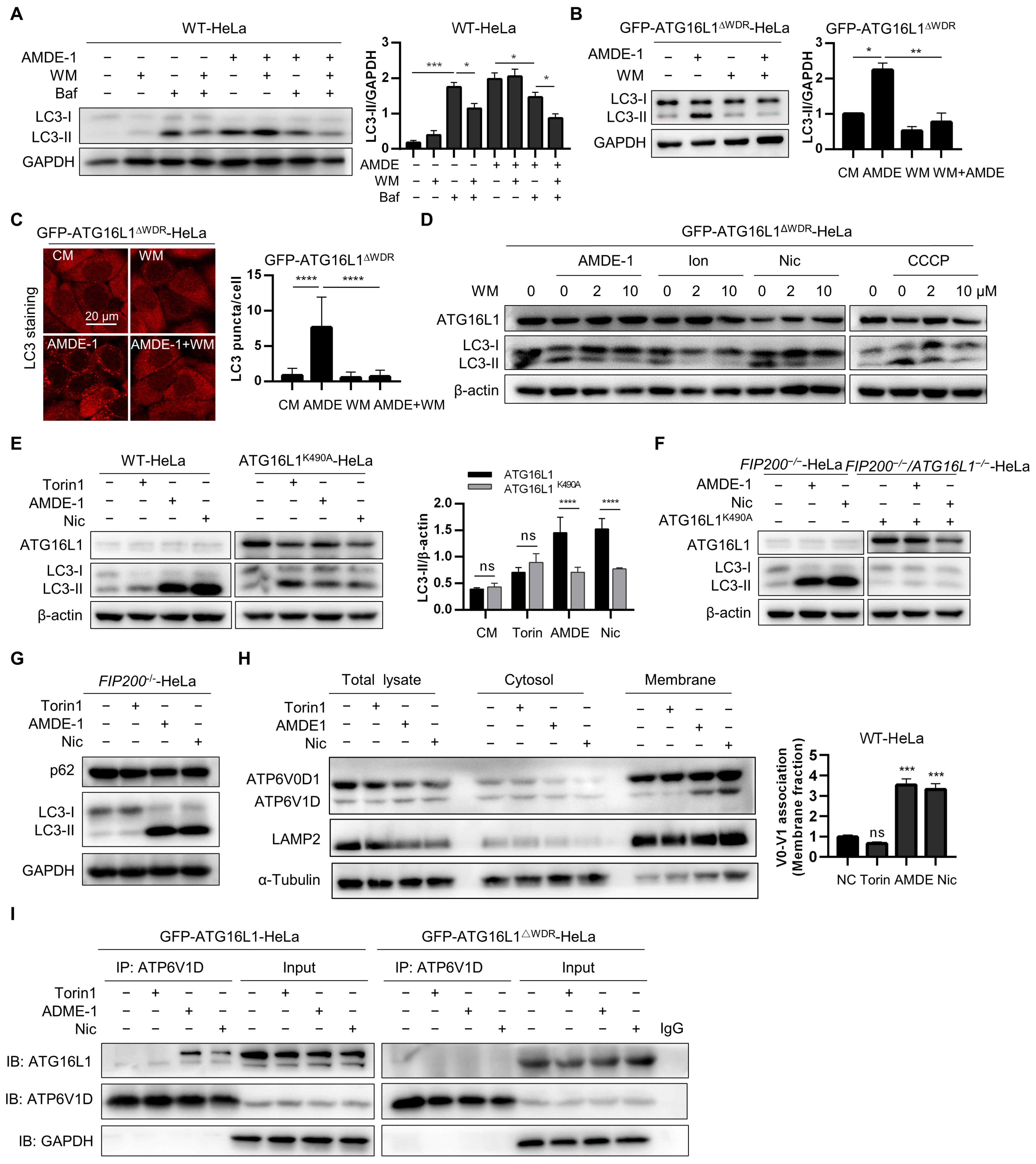
2.5. Deletion of the WDR Domain or Mutation of K490A of ATG16L1 Inhibits Chemical-Induced NCA via Suppressing V-ATPase-ATG16L1 Axis
2.6. Intact ATG16L1 with a Full-Length WDR Is Required for Canonical Autophagy and NCA
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Lines and Culture Conditions
4.3. Plasmid Construction
4.4. Plasmid Transfection
4.5. DQ-BSA Assay
4.6. Immunostaining
4.7. Immunoblotting
4.8. Immunoprecipitation Assay
4.9. LLP Degradation Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Overholtzer, M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012, 22, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Henault, J.; Martinez, J.; Riggs, J.M.; Tian, J.; Mehta, P.; Clarke, L.; Sasai, M.; Latz, E.; Brinkmann, M.M.; Iwasaki, A.; et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 2012, 37, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.L.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Lu, H.; Liao, L.; Li, S.; Xiong, H.; Yao, J. Bioengineered Nanospores Selectively Blocking LC3-Associated Phagocytosis in Tumor-Associated Macrophages Potentiate Antitumor Immunity. ACS Nano 2023, 17, 10872–10887. [Google Scholar] [CrossRef]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Hubber, A.; Kubori, T.; Coban, C.; Matsuzawa, T.; Ogawa, M.; Kawabata, T.; Yoshimori, T.; Nagai, H. Bacterial secretion system skews the fate of Legionella-containing vacuoles towards LC3-associated phagocytosis. Sci. Rep. 2017, 7, 44795. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Sønder, S.L.; Häger, S.C.; Heitmann, A.S.B.; Frankel, L.B.; Dias, C.; Simonsen, A.C.; Nylandsted, J. Restructuring of the plasma membrane upon damage by LC3-associated micropinocytosis. Sci. Adv. 2021, 7, eabg1969. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Magné, J.; Green, D.R. LC3-associated endocytosis and the functions of Rubicon and ATG16L1. Sci. Adv. 2022, 8, eabo5600. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ram, A.; Albeck, J.G.; Overholtzer, M. Entosis is induced by ultraviolet radiation. iScience 2021, 24, 102902. [Google Scholar] [CrossRef]
- de Duve, C.; de Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; Van Hoof, F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Marceau, F.; Bawolak, M.T.; Lodge, R.; Bouthillier, J.; Gagné-Henley, A.; Gaudreault, R.C.; Morissette, G. Cation trapping by cellular acidic compartments: Beyond the concept of lysosomotropic drugs. Toxicol. Appl. Pharmacol. 2012, 259, 1–12. [Google Scholar] [CrossRef]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef]
- Jacquin, E.; Leclerc-Mercier, S.; Judon, C.; Blanchard, E.; Fraitag, S.; Florey, O. Pharmacological modulators of autophagy activate a parallel noncanonical pathway driving unconventional LC3 lipidation. Autophagy 2017, 13, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.M.; Walkup WG 4th Hooper, K.; Li, T.; Kishi-Itakura, C.; Ng, A.; Lehmberg, T.; Jha, A.; Kommineni, S.; Fletcher, K.; Garcia-Fortanet, J.; et al. GABARAP sequesters the FLCN-FNIP tumor suppressor complex to couple autophagy with lysosomal biogenesis. Sci. Adv. 2021, 7, eabj2485. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, X.; Shan, H.; Fu, Y.; Gu, Q.; Zheng, X.; Dai, Q.; Xia, F.; Zheng, Z.; Liu, P.; et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells 2019, 8, 248. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Y.; Hong, L.; Yang, Z.; Cai, X.; Chen, X.; Fu, Y.; Lin, Y.; Wen, W.; Li, S.; et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 2016, 7, e2330. [Google Scholar] [CrossRef]
- Wang, Y.; Sharma, P.; Jefferson, M.; Zhang, W.; Bone, B.; Kipar, A.; Bitto, D.; Coombes, J.L.; Pearson, T.; Man, A.; et al. Non-canonical autophagy functions of ATG16L1 in epithelial cells limit lethal infection by influenza A virus. EMBO J. 2021, 40, e105543. [Google Scholar] [CrossRef]
- Fischer, T.D.; Wang, C.X.; Padman, B.S.; Lazarou, M.; Youle, R.J. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J. Cell Biol. 2020, 219, e202009128. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Huang, H.H.; Marsh, T.; Solvik, T.; Zhang, D.; Ye, J.; Kai, F.-B.; Goldsmith, J.; Liu, J.Y.; Huang, Y.-H.; et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 2020, 22, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Noda, N.N.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J. Biol. Chem. 2010, 285, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Bajagic, M.; Archna, A.; Busing, P.; Scrima, A. Structure of the WD40-domain of human ATG16L1. Protein Sci. 2017, 26, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Fujita, N.; Kanno, E.; Omori, H.; Yoshimori, T.; Itoh, T.; Fukuda, M. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12–5-16L2 complex. Autophagy 2011, 7, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Arasteh, M.; Jefferson, M.; Pearson, T.; Wang, Y.X.; Zhang, W.J.; Bicsak, B.; Divekar, D.; Powell, P.P.; Naumann, R.; et al. The ATG5-binding and coiled coil domains of ATG16L1 maintain autophagy and tissue homeostasis in mice independently of the WD domain required for LC3-associated phagocytosis. Autophagy 2019, 15, 599–612. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Teubner BJ, W.; Boada-Romero, E.; Tummers, B.; Guy, C.; Fitzgerald, P.; Mayer, U.; Carding, S.; Zakharenko, S.S.; Wileman, T.; et al. Noncanonical function of an autophagy protein prevents spontaneous Alzheimer’s disease. Sci. Adv. 2020, 6, eabb9036. [Google Scholar] [CrossRef]
- Hooper, K.M.; Jacquin, E.; Li TY, N.; Goodwin, J.M.; Brumell, J.H.; Durgan, J.; Florey, O. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J. Cell Biol. 2022, 221, e202105112. [Google Scholar] [CrossRef]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. Embo. J. 2018, 37, e97840. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, P.; Cheng, S.; Lu, Q.; Nowak, K.; Hopp, A.K.; Li, L.; Shi, X.; Zhou, Z.; Gao, W.; et al. A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis that Initiates Xenophagy. Cell 2019, 178, 552–566.e520. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; Chen, X.; Li, M.; Zhang, H.; Ding, W.X.; Yin, X.M. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem. Bioph. Res. Co. 2013, 432, 226–230. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaizuka, T.; Cadwell, K.; Sahani, M.H.; Saitoh, T.; Akira, S.; Virgin, H.W.; Mizushima, N. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013, 14, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Florey, O.; Overholtzer, M.; Jiang, X.J. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat. Struct. Mol. Biol. 2013, 20, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson HE, J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef]
- Li, M.; Yang, Z.L.; Vollmer, L.L.; Gao, Y.; Fu, Y.Y.; Liu, C.; Chen, X.Y.; Liu, P.Q.; Vogt, A.; Yin, X.M. AMDE-1 Is a Dual Function Chemical for Autophagy Activation and Inhibition. PLoS ONE 2015, 10, e0122083. [Google Scholar] [CrossRef]
- Ulferts, R.; Marcassa, E.; Timimi, L.; Lee, L.C.; Daley, A.; Montaner, B.; Turner, S.D.; Florey, O.; Baillie, J.K.; Beale, R. Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Rep. 2021, 37, 109899. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.P.; Forgac, M. Regulation and function of V-ATPases in physiology and disease. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183341. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Chen, Z.X.; Stang, M.T.; Gao, W.T. Transiently expressed ATG16L1 inhibits autophagosome biogenesis and aberrantly targets RAB11-positive recycling endosomes. Autophagy 2017, 13, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Saitoh, T.; Kageyama, S.; Akira, S.; Noda, T.; Yoshimori, T. Differential Involvement of Atg16L1 in Crohn Disease and Canonical Autophagy Analysis of the Organization of the Atg16L1 Complex in Fibroblasts. J. Biol. Chem. 2009, 284, 32602–32609. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Cristofani, R.; Marelli, M.M.; Cicardi, M.E.; Fontana, F.; Marzagalli, M.; Limonta, P.; Poletti, A.; Moretti, R.M. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018, 9, 889. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, G.; Leduc, M.; Muller, K.; Liu, P.; Zhao, L.W.; Humeau, J.; Xie, W.; Zhang, S.; Kepp, O.; Sauvat, A.; et al. Oleate-induced aggregation of LC3 at the trans-Golgi network is linked to a protein trafficking blockade. Cell Death Differ. 2021, 28, 1733–1752. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Lystad, A.H.; Sloan, K.; Carlsson, S.R.; Wilson, M.I.; Marcassa, E.; Ulferts, R.; Webster, J.; Lopez-Clavijo, A.F.; Wakelam, M.J.; et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 2021, 81, 2031. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.H.; Chen, X.Y.; Chen, D.H.; Vollmer, L.; Liu, P.Q.; Vogt, A.; Yin, X.M. Suppression of Lysosome Function Induces Autophagy via a Feedback Down-regulation of MTOR Complex 1 (MTORC1) Activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Lee, Y.M.; Ng, S.; Shi, Y.; Hua, Z.C.; Lin, Q.; Shen, H.M. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat. Protoc. 2017, 12, 279–288. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Fang, D.; Zhong, J.; Li, M. Missing WD40 Repeats in ATG16L1 Delays Canonical Autophagy and Inhibits Noncanonical Autophagy. Int. J. Mol. Sci. 2024, 25, 4493. https://doi.org/10.3390/ijms25084493
Tang J, Fang D, Zhong J, Li M. Missing WD40 Repeats in ATG16L1 Delays Canonical Autophagy and Inhibits Noncanonical Autophagy. International Journal of Molecular Sciences. 2024; 25(8):4493. https://doi.org/10.3390/ijms25084493
Chicago/Turabian StyleTang, Jiuge, Dongmei Fang, Jialing Zhong, and Min Li. 2024. "Missing WD40 Repeats in ATG16L1 Delays Canonical Autophagy and Inhibits Noncanonical Autophagy" International Journal of Molecular Sciences 25, no. 8: 4493. https://doi.org/10.3390/ijms25084493
APA StyleTang, J., Fang, D., Zhong, J., & Li, M. (2024). Missing WD40 Repeats in ATG16L1 Delays Canonical Autophagy and Inhibits Noncanonical Autophagy. International Journal of Molecular Sciences, 25(8), 4493. https://doi.org/10.3390/ijms25084493