
Triggering Degradation of Host Cellular Proteins for Robust Propagation of Influenza Viruses



Abstract
:1. Introduction
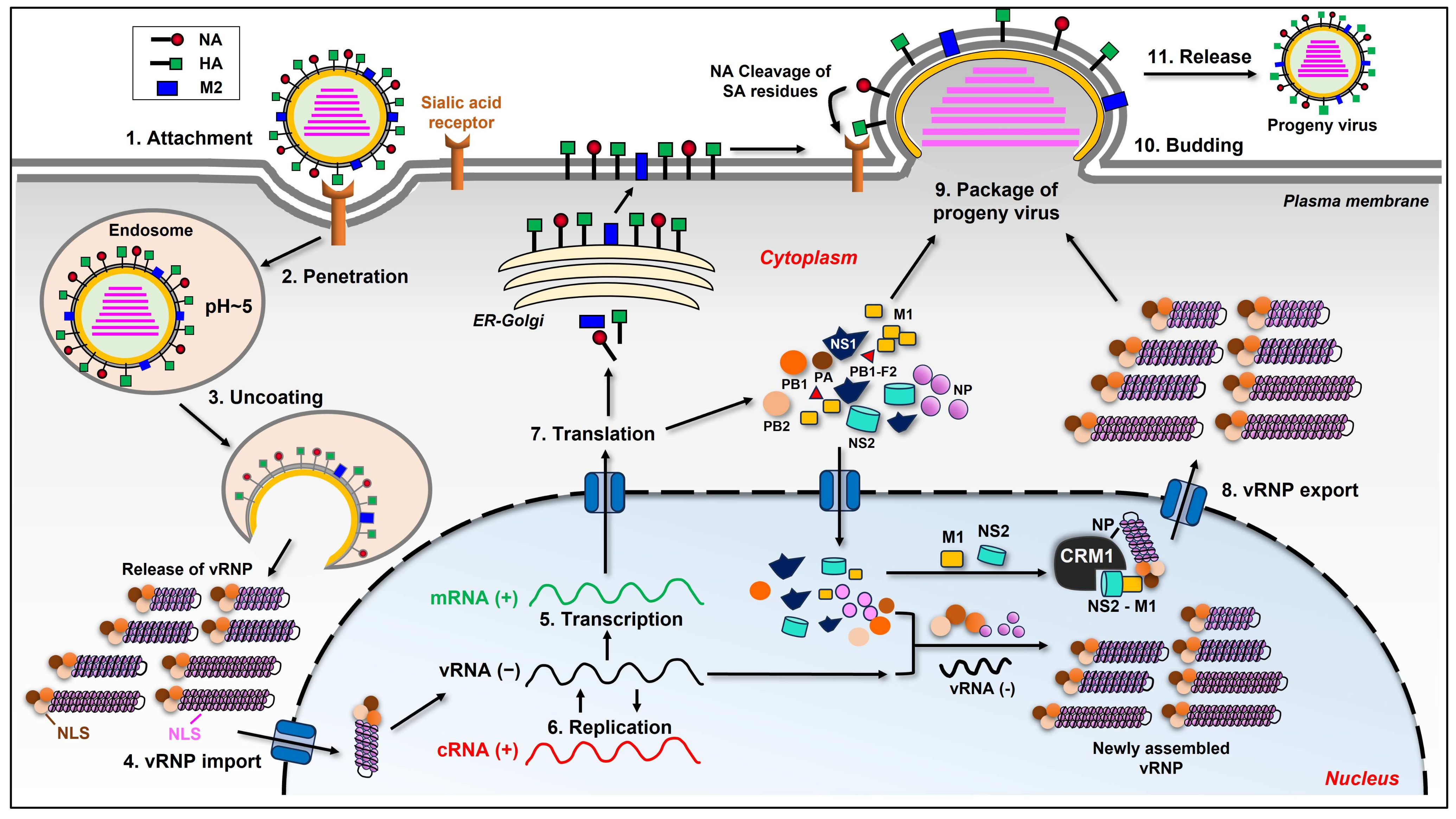
2. Influenza Virus Life Cycle
3. Influenza Viral Degradation of Cellular Proteins Involved in Interferon (IFN) Responses
3.1. Viral Degradation of Cellular Factors That Regulate Type I IFN Production Pathway
3.2. Viral Degradation of Proteins in the IFN-I Receptor Signaling
3.3. Viral Degradation of the Cellular Factors in the Type II and Type III IFN Pathways
4. Viral Degradation of Cellular Proteins That Do Not Directly Regulate IFN Responses
4.1. Viral RNA Polymerase
4.2. Viral NS1
4.3. Viral M2
4.4. Viral NS2 and NA
4.5. Yet-Unidentified Viral Component
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sutton, T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses 2018, 10, 461. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hou, S.; Xiong, C.; Hu, L.; Gong, L.; Yu, J.; Zhou, X.; Chen, Q.; Yuan, Y.; He, L.; et al. Avian influenza A virus H7N9 in China, a role reversal from reassortment receptor to the donator. J. Med. Virol. 2023, 95, e28392. [Google Scholar] [CrossRef] [PubMed]
- Lamotte, L.A.; Tafforeau, L. How Influenza A Virus NS1 Deals with the Ubiquitin System to Evade Innate Immunity. Viruses 2021, 13, 2309. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Wolff, T.; Ludwig, S. Activation of phosphatidylinositol 3-kinase signaling by the nonstructural NS1 protein is not conserved among type A and B influenza viruses. J. Virol. 2007, 81, 12097–12100. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Wang, X.; Ehrhardt, C.; Zheng, H.; Donelan, N.; Planz, O.; Pleschka, S.; Garcia-Sastre, A.; Heins, G.; Wolff, T. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J. Virol. 2002, 76, 11166–11171. [Google Scholar] [CrossRef] [PubMed]
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159. [Google Scholar] [PubMed]
- Dou, D.; Revol, R.; Ostbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Sheppard, C.M.; Staller, E.; Barclay, W.S. Host Determinants of Influenza RNA Synthesis. Annu. Rev. Virol. 2019, 6, 215–233. [Google Scholar] [CrossRef]
- Kamal, R.P.; Alymova, I.V.; York, I.A. Evolution and Virulence of Influenza A Virus Protein PB1-F2. Int. J. Mol. Sci. 2017, 19, 96. [Google Scholar] [CrossRef]
- Vreede, F.T.; Fodor, E. The role of the influenza virus RNA polymerase in host shut-off. Virulence 2010, 1, 436–439. [Google Scholar] [CrossRef]
- Zurcher, T.; Marion, R.M.; Ortin, J. Protein synthesis shut-off induced by influenza virus infection is independent of PKR activity. J. Virol. 2000, 74, 8781–8784. [Google Scholar] [CrossRef]
- Burzynska, P.; Sobala, L.F.; Mikolajczyk, K.; Jodlowska, M.; Jaskiewicz, E. Sialic Acids as Receptors for Pathogens. Biomolecules 2021, 11, 831. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Shinya, K.; Ebina, M.; Yamada, S.; Ono, M.; Kasai, N.; Kawaoka, Y. Avian flu: Influenza virus receptors in the human airway. Nature 2006, 440, 435–436. [Google Scholar] [CrossRef]
- Ma, W. Swine influenza virus: Current status and challenge. Virus Res. 2020, 288, 198118. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Kan, Q.; He, D.; Zhao, Z.; Gong, J.; Jiang, W.; Tang, T.; Li, Y.; Xie, Q.; Li, T.; et al. Phylogeography and Biological Characterizations of H12 Influenza A Viruses. Viruses 2022, 14, 2251. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Olivier, A.K.; Lu, X.; Epperson, W.; Zhang, X.; Zhong, L.; Waters, K.; Mamaliger, N.; Li, L.; Wen, F.; et al. The Sialyl Lewis X Glycan Receptor Facilitates Infection of Subtype H7 Avian Influenza A Viruses. J. Virol. 2022, 96, e0134422. [Google Scholar] [CrossRef]
- Broszeit, F.; Tzarum, N.; Zhu, X.Y.; Nemanichvili, N.; Eggink, D.; Leenders, T.; Li, Z.S.; Liu, L.; Wolfert, M.A.; Papanikolaou, A.; et al. N-Glycolylneuraminic Acid as a Receptor for Influenza A Viruses. Cell Rep. 2019, 27, 3284–3294.e6. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Boulo, S.; Akarsu, H.; Ruigrok, R.W.; Baudin, F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007, 124, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.G.; Fodor, E. Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 2006, 16, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.P.; Fodor, E. Interplay between Influenza Virus and the Host RNA Polymerase II Transcriptional Machinery. Trends Microbiol. 2019, 27, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Te Velthuis, A.J.W. Structure and Function of the Influenza Virus Transcription and Replication Machinery. Cold Spring Harb. Perspect. Med. 2020, 10, a038398. [Google Scholar] [CrossRef] [PubMed]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Muller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Hahm, B. Type I Interferon Modulates the Battle of Host Immune System Against Viruses. Adv. Appl. Microbiol. 2010, 73, 83–101. [Google Scholar] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011, 162, 12–18. [Google Scholar] [CrossRef]
- Hrincius, E.R.; Dierkes, R.; Anhlan, D.; Wixler, V.; Ludwig, S.; Ehrhardt, C. Phosphatidylinositol-3-kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig-I to promote efficient type I interferon production. Cell. Microbiol. 2011, 13, 1907–1919. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, S.; Liu, M.; Wei, Y.; Wang, Q.; Shen, W.; Lei, C.Q.; Zhu, Q. The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 2023, 19, 1916–1933. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Xu, S.; Wei, Y.; Zhang, X.; Wang, Q.; Jia, Y.; Wang, W.; Han, L.; Chen, Z.; Wang, Z.; et al. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021, 17, e1009300. [Google Scholar] [CrossRef]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef]
- Ouyang, W.; Cen, M.; Yang, L.; Zhang, W.; Xia, J.; Xu, F. NMI Facilitates Influenza A Virus Infection by Promoting Degradation of IRF7 through TRIM21. Am. J. Respir. Cell Mol. Biol. 2021, 65, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Blake, C.; Alexander, S.; Hahm, B. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J. Virol. 2010, 84, 8124–8131. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Xia, C.; Song, Y.E.; Ngo, H.; Studstill, C.J.; Drews, K.; Fox, T.E.; Johnson, M.C.; Hiscott, J.; Kester, M.; et al. Sphingosine 1-Phosphate Lyase Enhances the Activation of IKKepsilon To Promote Type I IFN-Mediated Innate Immune Responses to Influenza A Virus Infection. J. Immunol. 2017, 199, 677–687. [Google Scholar] [CrossRef]
- Wolf, J.J.; Xia, C.; Studstill, C.J.; Ngo, H.; Brody, S.L.; Anderson, P.E.; Hahm, B. Influenza A virus NS1 induces degradation of sphingosine 1-phosphate lyase to obstruct the host innate immune response. Virology 2021, 558, 67–75. [Google Scholar] [CrossRef]
- Wolf, J.J.; Saba, J.D.; Hahm, B. Analyzing Opposing Interactions Between Sphingosine 1-Phosphate Lyase and Influenza A Virus. DNA Cell Biol. 2022, 41, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Jahan, A.S.; Biquand, E.; Munoz-Moreno, R.; Le Quang, A.; Mok, C.K.; Wong, H.H.; Teo, Q.W.; Valkenburg, S.A.; Chin, A.W.H.; Man Poon, L.L.; et al. OTUB1 Is a Key Regulator of RIG-I-Dependent Immune Signaling and Is Targeted for Proteasomal Degradation by Influenza A NS1. Cell Rep. 2020, 30, 1570–1584.e6. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochem. Pharmacol. 2010, 79, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008, 27, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Fullam, A.; Schroder, M. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta 2013, 1829, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Fullam, A.; Brennan, R.; Schroder, M. Human DEAD box helicase 3 couples IkappaB kinase epsilon to interferon regulatory factor 3 activation. Mol. Cell. Biol. 2013, 33, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Byun, Y.H.; Park, S.; Jang, Y.H.; Han, W.R.; Won, J.; Cho, K.C.; Kim, D.H.; Lee, A.R.; Shin, G.C.; et al. Co-degradation of interferon signaling factor DDX3 by PB1-F2 as a basis for high virulence of 1918 pandemic influenza. Embo J. 2019, 38, e99475. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Campos, A.D.; Darnay, B.G.; Bentz, G.L.; Pagano, J.S. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 2008, 28, 6536–6546. [Google Scholar] [CrossRef]
- Lee, N.R.; Ban, J.; Lee, N.J.; Yi, C.M.; Choi, J.Y.; Kim, H.; Lee, J.K.; Seong, J.; Cho, N.H.; Jung, J.U.; et al. Activation of RIG-I-Mediated Antiviral Signaling Triggers Autophagy Through the MAVS-TRAF6-Beclin-1 Signaling Axis. Front. Immunol. 2018, 9, 2096. [Google Scholar] [CrossRef]
- Chen, Z.; Zeng, Y.; Wei, Y.; Wang, Q.; Liu, M.; Zhang, B.; Liu, J.; Zhu, Q.; Xu, S. Influenza D virus Matrix protein 1 restricts the type I interferon response by degrading TRAF6. Virology 2022, 568, 1–11. [Google Scholar] [CrossRef]
- Kash, J.C.; Tumpey, T.M.; Proll, S.C.; Carter, V.; Perwitasari, O.; Thomas, M.J.; Basler, C.F.; Palese, P.; Taubenberger, J.K.; Garcia-Sastre, A.; et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 2006, 443, 578–581. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Vijayan, M.; Pritzl, C.J.; Fuchs, S.Y.; McDermott, A.B.; Hahm, B. Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1. J. Virol. 2015, 90, 2403–2417. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Wolf, J.J.; Sun, C.; Xu, M.; Studstill, C.J.; Chen, J.; Ngo, H.; Zhu, H.; Hahm, B. PARP1 Enhances Influenza A Virus Propagation by Facilitating Degradation of Host Type I Interferon Receptor. J. Virol. 2020, 94, e01572-19. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Wolf, J.J.; Vijayan, M.; Studstill, C.J.; Ma, W.; Hahm, B. Casein Kinase 1alpha Mediates the Degradation of Receptors for Type I and Type II Interferons Caused by Hemagglutinin of Influenza A Virus. J. Virol. 2018, 92, e00006-18. [Google Scholar] [CrossRef]
- Yang, H.; Dong, Y.; Bian, Y.; Xu, N.; Wu, Y.; Yang, F.; Du, Y.; Qin, T.; Chen, S.; Peng, D.; et al. The influenza virus PB2 protein evades antiviral innate immunity by inhibiting JAK1/STAT signalling. Nat. Commun. 2022, 13, 6288. [Google Scholar] [CrossRef]
- Yang, H.; Dong, Y.; Bian, Y.; Huo, C.; Zhu, C.; Qin, T.; Chen, S.; Peng, D.; Liu, X. The synergistic effect of residues 32T and 550L in the PA protein of H5 subtype avian influenza virus contributes to viral pathogenicity in mice. PLoS Pathog. 2023, 19, e1011489. [Google Scholar] [CrossRef]
- Du, Y.; Yang, F.; Wang, Q.; Xu, N.; Xie, Y.; Chen, S.; Qin, T.; Peng, D. Influenza a virus antagonizes type I and type II interferon responses via SOCS1-dependent ubiquitination and degradation of JAK1. Virol. J. 2020, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, L.; Xu, G.; Cao, Z.; Wang, Q.; Li, S.; Peng, N.; Yin, J.; Yu, H.; Li, M.; et al. Epigenetic Modification Is Regulated by the Interaction of Influenza A Virus Nonstructural Protein 1 with the De Novo DNA Methyltransferase DNMT3B and Subsequent Transport to the Cytoplasm for K48-Linked Polyubiquitination. J. Virol. 2019, 93, e01587-18. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [PubMed]
- Liu, S.Y.; Sanchez, D.J.; Aliyari, R.; Lu, S.; Cheng, G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 2012, 109, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Rhein, B.A.; Powers, L.S.; Rogers, K.; Anantpadma, M.; Singh, B.K.; Sakurai, Y.; Bair, T.; Miller-Hunt, C.; Sinn, P.; Davey, R.A.; et al. Interferon-gamma Inhibits Ebola Virus Infection. PLoS Pathog. 2015, 11, e1005263. [Google Scholar] [CrossRef]
- Chaudhary, V.; Yuen, K.S.; Chan, J.F.; Chan, C.P.; Wang, P.H.; Cai, J.P.; Zhang, S.; Liang, M.; Kok, K.H.; Chan, C.P.; et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J. Virol. 2017, 91, e00163-17. [Google Scholar] [CrossRef]
- Brown, D.M.; Lee, S.; Garcia-Hernandez Mde, L.; Swain, S.L. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J. Virol. 2012, 86, 6792–6803. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Fan, L.; Du, H.; Xiang, B.; Li, Y.; Sun, M.; Kang, Y.; Chen, L.; Xu, C.; Li, Y.; et al. Recombinant Duck Interferon Gamma Inhibits H5N1 Influenza Virus Replication In Vitro and In Vivo. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2018, 38, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Reith, W.; LeibundGut-Landmann, S.; Waldburger, J.M. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 2005, 5, 793–806. [Google Scholar] [CrossRef]
- Ting, J.P.; Trowsdale, J. Genetic control of MHC class II expression. Cell 2002, 109 (Suppl. S21–S33). [Google Scholar] [CrossRef]
- Donnelly, R.P.; Kotenko, S.V. Interferon-lambda: A new addition to an old family. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2010, 30, 555–564. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef] [PubMed]
- Uetani, K.; Hiroi, M.; Meguro, T.; Ogawa, H.; Kamisako, T.; Ohmori, Y.; Erzurum, S.C. Influenza A virus abrogates IFN-gamma response in respiratory epithelial cells by disruption of the Jak/Stat pathway. Eur. J. Immunol. 2008, 38, 1559–1573. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.; Osman, W.; Adair, J.; ElMergawy, R.; Chafin, L.; Johns, F.; Farkas, D.; Elhance, A.; Londino, J.; Mallampalli, R.K. The E3 ligase subunit FBXO45 binds the interferon-lambda receptor and promotes its degradation during influenza virus infection. J. Biol. Chem. 2022, 298, 102698. [Google Scholar] [CrossRef] [PubMed]
- Vreede, F.T.; Chan, A.Y.; Sharps, J.; Fodor, E. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 2010, 396, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Llompart, C.M.; Nieto, A.; Rodriguez-Frandsen, A. Specific residues of PB2 and PA influenza virus polymerase subunits confer the ability for RNA polymerase II degradation and virus pathogenicity in mice. J. Virol. 2014, 88, 3455–3463. [Google Scholar] [CrossRef]
- Martinez-Alonso, M.; Hengrung, N.; Fodor, E. RNA-Free and Ribonucleoprotein-Associated Influenza Virus Polymerases Directly Bind the Serine-5-Phosphorylated Carboxyl-Terminal Domain of Host RNA Polymerase II. J. Virol. 2016, 90, 6014–6021. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Perez-Gonzalez, A.; Nieto, A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J. Virol. 2007, 81, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Perez-Gonzalez, A.; Hossain, M.J.; Chen, L.M.; Rolling, T.; Perez-Brena, P.; Donis, R.; Kochs, G.; Nieto, A. Attenuated strains of influenza A viruses do not induce degradation of RNA polymerase II. J. Virol. 2009, 83, 11166–11174. [Google Scholar] [CrossRef]
- Alfonso, R.; Rodriguez, A.; Rodriguez, P.; Lutz, T.; Nieto, A. CHD6, a cellular repressor of influenza virus replication, is degraded in human alveolar epithelial cells and mice lungs during infection. J. Virol. 2013, 87, 4534–4544. [Google Scholar] [CrossRef]
- Alfonso, R.; Lutz, T.; Rodriguez, A.; Chavez, J.P.; Rodriguez, P.; Gutierrez, S.; Nieto, A. CHD6 chromatin remodeler is a negative modulator of influenza virus replication that relocates to inactive chromatin upon infection. Cell. Microbiol. 2011, 13, 1894–1906. [Google Scholar] [CrossRef]
- Nagesh, P.T.; Husain, M. Influenza A Virus Dysregulates Host Histone Deacetylase 1 That Inhibits Viral Infection in Lung Epithelial Cells. J. Virol. 2016, 90, 4614–4625. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.T.; Hussain, M.; Galvin, H.D.; Husain, M. Histone Deacetylase 2 Is a Component of Influenza A Virus-Induced Host Antiviral Response. Front. Microbiol. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed]
- Galvin, H.D.; Husain, M. Influenza A virus-induced host caspase and viral PA-X antagonize the antiviral host factor, histone deacetylase 4. J. Biol. Chem. 2019, 294, 20207–20221. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Cheung, C.Y. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Nutsford, A.N.; Galvin, H.D.; Ahmed, F.; Husain, M. The Class IV human deacetylase, HDAC11, exhibits anti-influenza A virus properties via its involvement in host innate antiviral response. Cell. Microbiol. 2019, 21, e12989. [Google Scholar] [CrossRef] [PubMed]
- Zanin, M.; DeBeauchamp, J.; Vangala, G.; Webby, R.J.; Husain, M. Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection. Viruses 2020, 12, 728. [Google Scholar] [CrossRef]
- Chen, H.; Qian, Y.; Chen, X.; Ruan, Z.; Ye, Y.; Chen, H.; Babiuk, L.A.; Jung, Y.S.; Dai, J. HDAC6 Restricts Influenza A Virus by Deacetylation of the RNA Polymerase PA Subunit. J. Virol. 2019, 93, e01896-18. [Google Scholar] [CrossRef]
- Hussain, M.; Ahmed, F.; Henzeler, B.; Husain, M. Anti-microbial host factor HDAC6 is antagonised by the influenza A virus through host caspases and viral PA. Febs J. 2023, 290, 2744–2759. [Google Scholar] [CrossRef]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef]
- Garcia-Sastre, A.; Biron, C.A. Type 1 interferons and the virus-host relationship: A lesson in detente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef]
- Wang, S.; Chi, X.; Wei, H.; Chen, Y.; Chen, Z.; Huang, S.; Chen, J.L. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J. Virol. 2014, 88, 8375–8385. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Sun, W.; Li, S.; Yang, J.; Yang, L.; Quan, G.; Gao, X.; Wang, Z.; Cheng, X.; Li, Z.; et al. Proteomics Analysis of Cellular Proteins Co-Immunoprecipitated with Nucleoprotein of Influenza A Virus (H7N9). Int. J. Mol. Sci. 2015, 16, 25982–25998. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, S.; Xu, F.; Mei, S.; Liu, X.; Yin, L.; Zhao, F.; Zhao, X.; Sun, H.; Xiong, Z.; et al. MOV10 sequesters the RNP of influenza A virus in the cytoplasm and is antagonized by viral NS1 protein. Biochem. J. 2019, 476, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Villalon-Letelier, F.; Reading, P.C. Unraveling the role of the MOV10 RNA helicase during influenza A virus infection. Biochem. J. 2019, 476, 1005–1008. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C.H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Lane, D.; Levine, A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef]
- Zhirnov, O.P.; Klenk, H.D. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis Int. J. Program. Cell Death 2007, 12, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Pazos, M.; Delgado, I.; Murk, W.; Mungamuri, S.K.; Lee, S.W.; García-Sastre, A.; Moran, T.M.; Aaronson, S.A. p53 Serves as a Host Antiviral Factor That Enhances Innate and Adaptive Immune Responses to Influenza A Virus. J. Immunol. 2011, 187, 6428–6436. [Google Scholar] [CrossRef]
- Taura, M.; Eguma, A.; Suico, M.A.; Shuto, T.; Koga, T.; Komatsu, K.; Komune, T.; Sato, T.; Saya, H.; Li, J.D.; et al. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol. Cell. Biol. 2008, 28, 6557–6567. [Google Scholar] [CrossRef]
- Yanai, H.; Chen, H.M.; Inuzuka, T.; Kondo, S.; Mak, T.W.; Takaoka, A.; Honda, K.; Taniguchi, T. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. USA 2007, 104, 3402–3407. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Lee, E.S.; Lim, D.S.; Bae, Y.S. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc. Natl. Acad. Sci. USA 2009, 106, 7852–7857. [Google Scholar] [CrossRef] [PubMed]
- Hummer, B.T.; Li, X.L.; Hassel, B.A. Role for p53 in gene induction by double-stranded RNA. J. Virol. 2001, 75, 7774–7777. [Google Scholar] [CrossRef] [PubMed]
- Hacke, K.; Rincon-Orozco, B.; Buchwalter, G.; Siehler, S.Y.; Wasylyk, B.; Wiesmuller, L.; Rosl, F. Regulation of MCP-1 chemokine transcription by p53. Mol. Cancer 2010, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Terrier, O.; Josset, L.; Textoris, J.; Marcel, V.; Cartet, G.; Ferraris, O.; N’Guyen, C.; Lina, B.; Diaz, J.J.; Bourdon, J.C.; et al. Cellular transcriptional profiling in human lung epithelial cells infected by different subtypes of influenza A viruses reveals an overall down-regulation of the host p53 pathway. Virol. J. 2011, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Ponnuswamy, A.; Hupp, T.; Fahraeus, R. Concepts in MDM2 Signaling: Allosteric Regulation and Feedback Loops. Genes Cancer 2012, 3, 291–297. [Google Scholar] [CrossRef]
- Wang, X.; Deng, X.; Yan, W.; Zhu, Z.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Wei, J.; Xia, X.; et al. Stabilization of p53 in influenza A virus-infected cells is associated with compromised MDM2-mediated ubiquitination of p53. J. Biol. Chem. 2012, 287, 18366–18375. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, A.; Dubois, J.; Machado, D.; Cartet, G.; Traversier, A.; Julien, T.; Lina, B.; Bourdon, J.C.; Rosa-Calatrava, M.; Terrier, O. Influenza A viruses alter the stability and antiviral contribution of host E3-ubiquitin ligase Mdm2 during the time-course of infection. Sci. Rep. 2018, 8, 3746. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, F.; Zou, Z.; Bi, Y.; Yang, Y.; Zhang, C.; Liu, Q.; Shang, D.; Yan, Y.; Ju, X.; et al. Avian influenza viruses suppress innate immunity by inducing trans-transcriptional readthrough via SSU72. Cell. Mol. Immunol. 2022, 19, 702–714. [Google Scholar] [CrossRef]
- Das, U.; Chawla-Sarkar, M.; Gangopadhyay, S.R.; Dey, S.; Sharma, R.D. Role of Influenza A virus protein NS1 in regulating host nuclear body ND10 complex formation and its involvement in establishment of viral pathogenesis. PLoS ONE 2024, 19, e0295522. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Nerome, K.; Moriyama, M.; Hayakawa, S.; Kuroda, K. Addition of an EGFP-tag to the N-terminal of influenza virus M1 protein impairs its ability to accumulate in ND10. J. Virol. Methods 2018, 252, 75–79. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshioka, K.; Suzuki, C.; Awashima, S.; Hosaka, Y.; Yewdell, J.; Kuroda, K. Localization of influenza virus proteins to nuclear dot 10 structures in influenza virus-infected cells. Virology 2003, 310, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Londino, J.D.; Lazrak, A.; Noah, J.W.; Aggarwal, S.; Bali, V.; Woodworth, B.A.; Bebok, Z.; Matalon, S. Influenza virus M2 targets cystic fibrosis transmembrane conductance regulator for lysosomal degradation during viral infection. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 2712–2725. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mok, C.K.; Chan, M.C.; Zhang, Y.; Nal, B.; Kien, F.; Bruzzone, R.; Sanyal, S. Cell Cycle-independent Role of Cyclin D3 in Host Restriction of Influenza Virus Infection. J. Biol. Chem. 2017, 292, 5070–5088. [Google Scholar] [CrossRef] [PubMed]
- Dreer, M.; Fertey, J.; van de Poel, S.; Straub, E.; Madlung, J.; Macek, B.; Iftner, T.; Stubenrauch, F. Interaction of NCOR/SMRT Repressor Complexes with Papillomavirus E8;E2C Proteins Inhibits Viral Replication. PLoS Pathog. 2016, 12, e1005556. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Kao, H.Y. G protein pathway suppressor 2 (GPS2) is a transcriptional corepressor important for estrogen receptor alpha-mediated transcriptional regulation. J. Biol. Chem. 2009, 284, 36395–36404. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; He, X.; Huang, K.; Zhang, Y.; Li, C.; Yang, Y.; Zou, Z.; Jin, M. Interaction of NEP with G Protein Pathway Suppressor 2 Facilitates Influenza A Virus Replication by Weakening the Inhibition of GPS2 to RNA Synthesis and Ribonucleoprotein Assembly. J. Virol. 2021, 95, e00008-21. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Yan, Y.; Liu, Q.; Li, N.; Sheng, M.; Zhang, L.; Li, X.; Liang, Z.; Huang, F.; Liu, K.; et al. Neuraminidase of Influenza A Virus Binds Lysosome-Associated Membrane Proteins Directly and Induces Lysosome Rupture. J. Virol. 2015, 89, 10347–10358. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, N.; Tang, J.; Liu, S.; Luo, D.; Duan, Q.; Wang, X. Downregulation of angiotensin-converting enzyme 2 by the neuraminidase protein of influenza A (H1N1) virus. Virus Res. 2014, 185, 64–71. [Google Scholar] [CrossRef]
- Yu, K.; Ren, Y.; Zhang, X.; Qiao, T.; Liu, Z.; Shi, J.; Wang, Y. shRNA-mediated NP knockdown inhibits the apoptosis of cardiomyocytes induced by H1N1pdm2009 influenza virus. Mol. Med. Rep. 2017, 16, 1376–1382. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Chen, P.; Tong, G.; Ma, Z. The non-structural (NS1) protein of influenza A virus associates with p53 and inhibits p53-mediated transcriptional activity and apoptosis. Biochem. Biophys. Res. Commun. 2010, 395, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix (M1) protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef]
- Krumbholz, A.; Philipps, A.; Oehring, H.; Schwarzer, K.; Eitner, A.; Wutzler, P.; Zell, R. Current knowledge on PB1-F2 of influenza A viruses. Med. Microbiol. Immunol. 2011, 200, 69–75. [Google Scholar] [CrossRef]
- Lowy, R.J. Influenza virus induction of apoptosis by intrinsic and extrinsic mechanisms. Int. Rev. Immunol. 2003, 22, 425–449. [Google Scholar] [CrossRef] [PubMed]
- Ampomah, P.B.; Lim, L.H.K. Influenza A virus-induced apoptosis and virus propagation. Apoptosis Int. J. Program. Cell Death 2020, 25, 1–11. [Google Scholar] [CrossRef]
- Divangahi, M.; King, I.L.; Pernet, E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol. 2015, 36, 307–314. [Google Scholar] [CrossRef]
- Jaworska, J.; Coulombe, F.; Downey, J.; Tzelepis, F.; Shalaby, K.; Tattoli, I.; Berube, J.; Rousseau, S.; Martin, J.G.; Girardin, S.E.; et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc. Natl. Acad. Sci. USA 2014, 111, E2110–E2119. [Google Scholar] [CrossRef]
- Cen, M.; Ouyang, W.; Lin, X.; Du, X.; Hu, H.; Lu, H.; Zhang, W.; Xia, J.; Qin, X.; Xu, F. FBXO6 regulates the antiviral immune responses via mediating alveolar macrophages survival. J. Med. Virol. 2023, 95, e28203. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Kroeze, E.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet. Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Ruan, T.; Sun, Y.L.; Zhang, J.T.; Sun, J.; Liu, W.; Prinz, R.A.; Peng, D.X.; Liu, X.F.; Xu, X.L. H5N1 infection impairs the alveolar epithelial barrier through intercellular junction proteins via Itch-mediated proteasomal degradation. Commun. Biol. 2022, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Husain, M. Caspase-Mediated Cleavage of Human Cortactin during Influenza A Virus Infection Occurs in Its Actin-Binding Domains and Is Associated with Released Virus Titres. Viruses 2020, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Husain, M. Caspase-mediated degradation of host cortactin that promotes influenza A virus infection in epithelial cells. Virology 2016, 497, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free Radic. Biol. Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]
- Pyo, C.W.; Shin, N.; Jung, K.I.; Choi, J.H.; Choi, S.Y. Alteration of copper-zinc superoxide dismutase 1 expression by influenza A virus is correlated with virus replication. Biochem. Biophys. Res. Commun. 2014, 450, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Pyo, C.W.; Choi, S.Y. Influenza A virus-induced autophagy contributes to enhancement of virus infectivity by SOD1 downregulation in alveolar epithelial cells. Biochem. Biophys. Res. Commun. 2018, 498, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Loutfy, M.R.; Blatt, L.M.; Siminovitch, K.A.; Ward, S.; Wolff, B.; Lho, H.; Pham, D.H.; Deif, H.; LaMere, E.A.; Chang, M.; et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: A preliminary study. JAMA 2003, 290, 3222–3228. [Google Scholar] [CrossRef]
- Debing, Y.; Neyts, J. Antiviral strategies for hepatitis E virus. Antivir. Res. 2014, 102, 106–118. [Google Scholar] [CrossRef]
- Jung, K.I.; McKenna, S.; Vijayamahantesh, V.; He, Y.; Hahm, B. Protective versus Pathogenic Type I Interferon Responses during Virus Infections. Viruses 2023, 15, 1916. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.; Yamauchi, Y. Ubiquitin in Influenza Virus Entry and Innate Immunity. Viruses 2016, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Dezhbord, M.; Lee, A.R.; Kim, K.H. The Roles of Ubiquitination in Pathogenesis of Influenza Virus Infection. Int. J. Mol. Sci. 2022, 23, 4593. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cellular Proteins | Viral Component | Function of the Cellular Proteins | Degradation Pathway | Relevant E3 Ubiquitin Ligase | Reference |
|---|---|---|---|---|---|
| OTUB1 | NS1 | Activates RIG-I via hydrolyzing K48-linked polyubiquitin chains of RIG-I | Proteasomal pathway | ND | [42] |
| MAVS | NP, PB1 | A key adaptor for transducing RIG-I mediated signaling | Mitophagy pathway | RNF5 | [33,34] |
| IRF7 | ND | A transcription factor that is activated downstream TBK1/IKKε and promotes the induction of type I IFNs | Proteasomal pathway | TRIM21 | [37] |
| SPL | NS1 | Interacts with IKKε to promote the IAV induced type I IFN response | Proteasomal pathway | ND | [38,39,40,41] |
| DDX3 | PB1-F2 | Interacts with IKKε and TBK1 and contributes to the induction of type I IFNs | Proteasomal pathway | ND | [43,44,45,46,47] |
| TRAF6 | M1 (IDV) | Interacts with MAVS and is involved in the activation of IRF3/IRF7 | Proteasomal pathway | KEAP1 | [48,49,50] |
| IFNAR1 | HA | Binds to IFN-α/β and mediates the type I IFN antiviral signaling | Proteasomal and lysosomal pathway | SCF (HOS) | [53,54,55] |
| JAK1 | PB2, PA (H5) | Transduces signal form diverse receptors and activates STAT1/2 | Proteasomal pathway | ND | [56,57,58] |
| DNMT3B | NS1 | Catalyzes DNA methylation affecting the expression of JAK-STAT regulators | Proteasomal pathway | ND | [59] |
| IFNGR1 | HA | Binds to IFN-γ and mediates the type II IFN signaling pathway | Lysosomal pathway | ND | [55] |
| IFNLR1 | ND | Binds to IFN-λ and mediates the type III IFN signaling pathway | Proteasomal pathway | FBXO45 | [73] |
| RNA polymerase II | PA, PB2 | An enzyme responsible for the transcription of mRNAs | ND | ND | [74,75,76,77,78] |
| CHD6 | PA, PB1, PB2 | Affects the binding of transcription factors thus modulating the initiation and elongation steps of transcription | ND | ND | [79,80] |
| HDAC1 | ND | The most abundant member of the class I HDACs in pulmonary endothelial cells, regulating diverse cellular procedures | Proteasomal pathway | ND | [81] |
| HDAC2 | ND | A class I HDAC that mostly targets histone H3 and H4 for deacetylation and plays important roles in multiple cellular events | Proteasomal pathway | ND | [82] |
| HDAC4 | PA-X | A member of class IIa HDACs that is localized to both the nucleus and cytoplasm and has no deacetylase activity | Cleaved by lysosome-associated caspase | ND | [83] |
| HDAC6 | PA | A class IIb HDAC that is non-nuclear and deacetylates cytoplasmic substrates such as tubulin. | Cleaved by lysosome-associated caspase | ND | [84] [86,87,88] |
| elF4B | NS1 | Regulates mRNA translation initiation | Lysosomal pathway | ND | [89,90,91] |
| MOV10 | NS1 | Interacts with influenza viral NP and sequesters the vRNP in the cytoplasm | Lysosomal pathway | ND | [92,93,94,95] |
| MDM2 | NS1 | Negatively regulates p53 pathway by promoting the ubiquitination and degradation of p53 | Proteasomal pathway | ND | [106,107,108,109] |
| SSU72 | NS1 | Involved in transcription termination process | ND | ND | [110] |
| ND-10 | NS1 | Involved in the replication of numerous viruses and in host cell responses to antiviral cytokines | SUMOylation mediated disruption | ND | [111] |
| CFTR | M2 | Influences cytokine responses and plays an important role in viral infection-associated inflammation and mortality | Lysosomal pathway | ND | [114] |
| cyclin D3 | M2 | A key regulator of cell cycle | Proteasomal pathway | ND | [115] |
| LAMPs | NA | Present in the lysosome membrane and function to maintain the structural integrity of lysosomal compartment to prevent hydrolytic enzyme release | Deglycosylation mediated diminishment | ND | [119] |
| ACE2 | NA | Serves as a functional receptor for both SARS-CoV and SARS-CoV-2 | Proteasomal pathway | ND | [120,121,122] |
| GPS2 | NS2 | Participates in the RAS/MAPK pathway and displays inhibitory function in cellular or viral transcription | Proteasomal pathway | ND | [116,117,118] |
| NLRX1 | ND | A pattern-recognition receptor belonging to the NLR family that localizes to the mitochondria | Proteasomal pathway | FBXO6 | [130,131] |
| occludin | ND | One of the major components of the tight junction in epithelial and endothelial cells which is crucial for the alveolar epithelial barrier | Proteasomal pathway | Itch | [133] |
| cortactin | ND | A central regulator of branched filamentous actin network and is associated with the infection of various bacterial and viral pathogens | Lysosomal pathway | ND | [134,135] |
| SOD1 | ND | An antioxidant enzyme that regulates cellular oxidative stress | Lysosomal pathway | ND | [136,137,138] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, C.; Wang, T.; Hahm, B. Triggering Degradation of Host Cellular Proteins for Robust Propagation of Influenza Viruses. Int. J. Mol. Sci. 2024, 25, 4677. https://doi.org/10.3390/ijms25094677
Xia C, Wang T, Hahm B. Triggering Degradation of Host Cellular Proteins for Robust Propagation of Influenza Viruses. International Journal of Molecular Sciences. 2024; 25(9):4677. https://doi.org/10.3390/ijms25094677
Chicago/Turabian StyleXia, Chuan, Ting Wang, and Bumsuk Hahm. 2024. "Triggering Degradation of Host Cellular Proteins for Robust Propagation of Influenza Viruses" International Journal of Molecular Sciences 25, no. 9: 4677. https://doi.org/10.3390/ijms25094677
APA StyleXia, C., Wang, T., & Hahm, B. (2024). Triggering Degradation of Host Cellular Proteins for Robust Propagation of Influenza Viruses. International Journal of Molecular Sciences, 25(9), 4677. https://doi.org/10.3390/ijms25094677