

Variable Penetrance and Expressivity of a Rare Pore Loss-of-Function Mutation (p.L889V) of Nav1.5 Channels in Three Spanish Families
 , , , ,
, , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clinical Description of Probands and Their Families
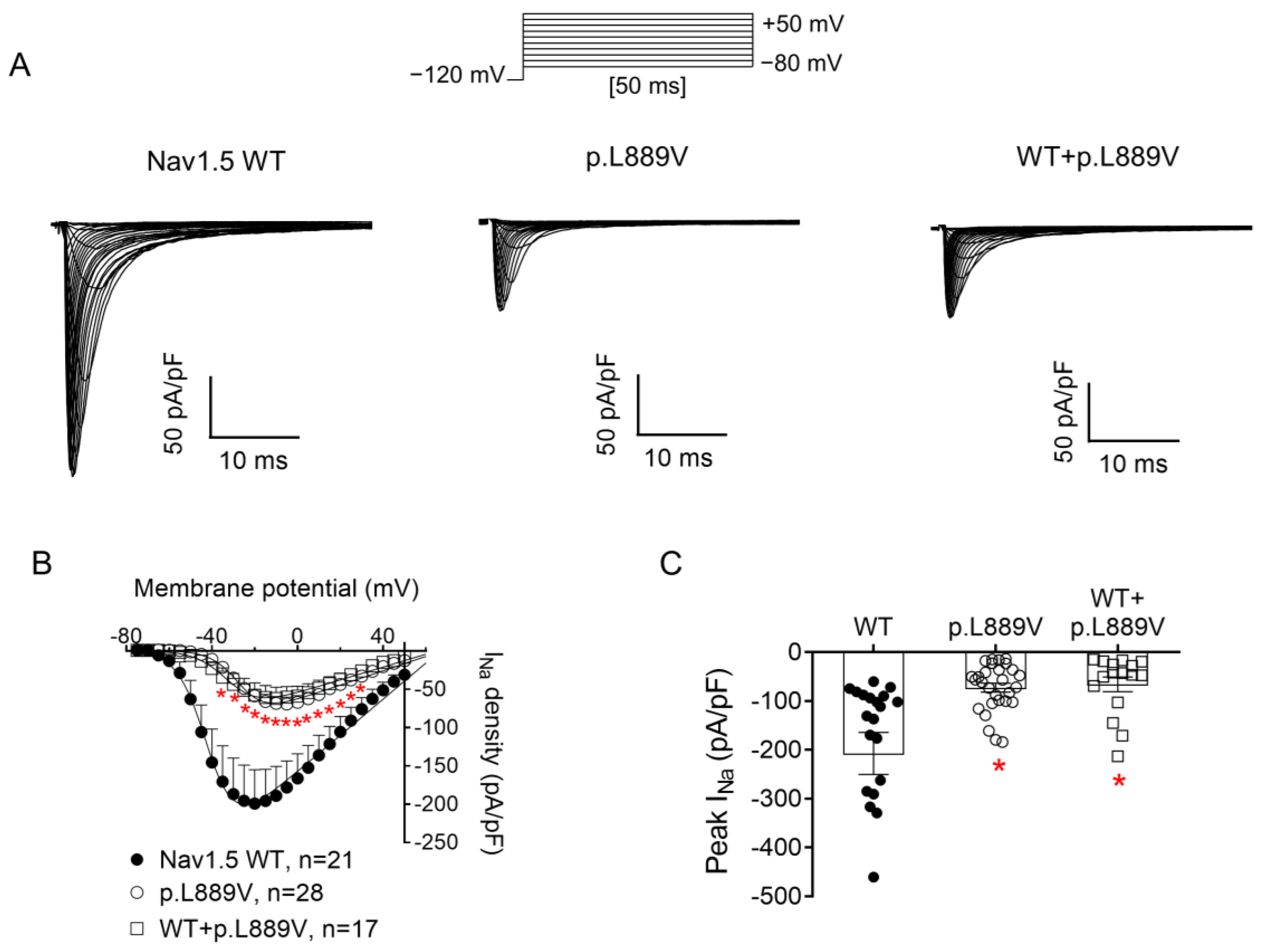
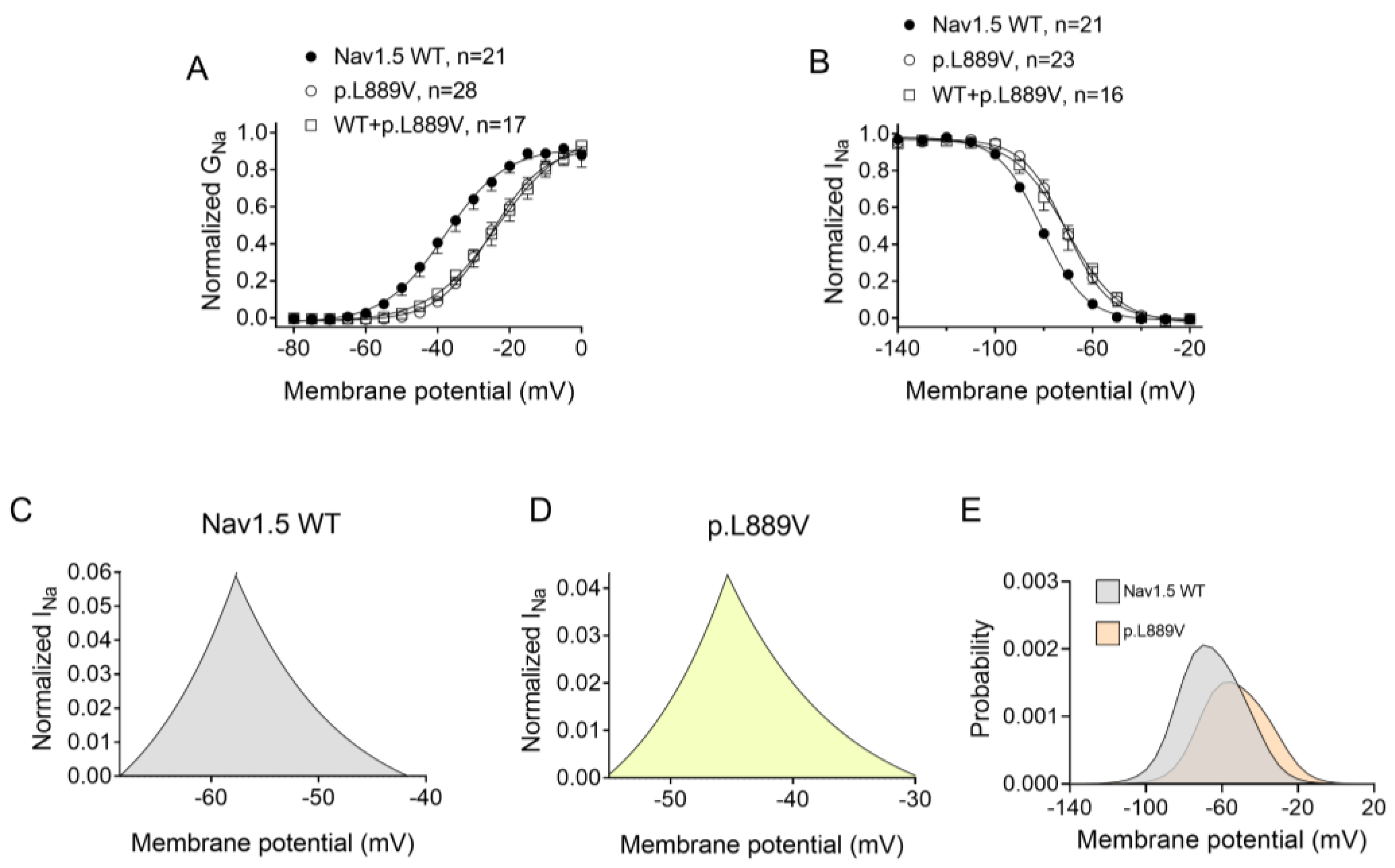
2.2. Functional Analysis of p.L889V Nav1.5 Channels
2.3. Membrane Expression of p.L889V Channels
2.4. Molecular Modeling
3. Discussion
Clinical Implications
4. Methods
4.1. Study Approval
4.2. Clinical Study and Genetic Analysis
4.3. cDNA Constructs, Mutagenesis, Cell Culture, and Transfection
4.4. Recording Techniques
4.5. Biotinylation Assay
4.6. Molecular Modeling
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Remme, C.A.; Verkerk, A.O.; Hoogaars, W.M.H.; Aanhaanen, W.T.J.; Scicluna, B.P.; Annink, C.; van den Hoff, M.J.B.; Wilde, A.A.; van Veen, T.A.B.; Veldkamp, M.W.; et al. The Cardiac Sodium Channel Displays Differential Distribution in the Conduction System and Transmural Heterogeneity in the Murine Ventricular Myocardium. Basic Res. Cardiol. 2009, 104, 511–522. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable Arrhythmia Syndromes Associated with Abnormal Cardiac Sodium Channel Function: Ionic and Non-Ionic Mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited Cardiac Arrhythmias. Nat. Rev. Dis. Primers 2020, 6, 58. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Remme, C.A.; Verkerk, A.O.; Nuyens, D.; van Ginneken, A.C.G.; van Brunschot, S.; Belterman, C.N.W.; Wilders, R.; van Roon, M.A.; Tan, H.L.; Wilde, A.A.M.; et al. Overlap Syndrome of Cardiac Sodium Channel Disease in Mice Carrying the Equivalent Mutation of Human SCN5A-1795insD. Circulation 2006, 114, 2584–2594. [Google Scholar] [CrossRef]
- Olson, T.M.; Keating, M.T. Mapping a Cardiomyopathy Locus to Chromosome 3p22-P25. J. Clin. Investig. 1996, 97, 528–532. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ciconte, G.; Monasky, M.M.; Santinelli, V.; Micaglio, E.; Vicedomini, G.; Anastasia, L.; Negro, G.; Borrelli, V.; Giannelli, L.; Santini, F.; et al. Brugada Syndrome Genetics Is Associated with Phenotype Severity. Eur. Heart J. 2021, 42, 1082–1090. [Google Scholar] [CrossRef]
- Glazer, A.M.; Wada, Y.; Li, B.; Muhammad, A.; Kalash, O.R.; O’Neill, M.J.; Shields, T.; Hall, L.; Short, L.; Blair, M.A.; et al. High-Throughput Reclassification of SCN5A Variants. Am. J. Hum. Genet. 2020, 107, 111–123. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage Gated Sodium and Calcium Channels: Discovery, Structure, Function, and Pharmacology. Channels 2023, 17, 2281714. [Google Scholar] [CrossRef]
- Crespo-García, T.; Rubio-Alarcón, M.; Cámara-Checa, A.; Dago, M.; Rapún, J.; Nieto-Marín, P.; Marín, M.; Cebrián, J.; Tamargo, J.; Delpón, E.; et al. A Cantú Syndrome Mutation Produces Dual Effects on KATP Channels by Disrupting Ankyrin B Regulation. J. Gen. Physiol. 2023, 155, e202112995. [Google Scholar] [CrossRef]
- Jiang, D.; Shi, H.; Tonggu, L.; Gamal El-Din, T.M.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Kroncke, B.M.; Glazer, A.M.; Smith, D.K.; Blume, J.D.; Roden, D.M. SCN5A (NaV1.5) Variant Functional Perturbation and Clinical Presentation: Variants of a Certain Significance. Circ. Genom. Precis. Med. 2018, 11, e002095. [Google Scholar] [CrossRef]
- Núñez, L.; Barana, A.; Amorós, I.; de la Fuente, M.G.; Dolz-Gaitón, P.; Gómez, R.; Rodríguez-García, I.; Mosquera, I.; Monserrat, L.; Delpón, E.; et al. P.D1690N Nav1.5 Rescues p.G1748D Mutation Gating Defects in a Compound Heterozygous Brugada Syndrome Patient. Heart Rhythm 2013, 10, 264–272. [Google Scholar] [CrossRef]
- Clatot, J.; Ziyadeh-Isleem, A.; Maugenre, S.; Denjoy, I.; Liu, H.; Dilanian, G.; Hatem, S.N.; Deschênes, I.; Coulombe, A.; Guicheney, P.; et al. Dominant-Negative Effect of SCN5A N-Terminal Mutations through the Interaction of Na(v)1.5 α-Subunits. Cardiovasc. Res. 2012, 96, 53–63. [Google Scholar] [CrossRef]
- Clatot, J.; Hoshi, M.; Wan, X.; Liu, H.; Jain, A.; Shinlapawittayatorn, K.; Marionneau, C.; Ficker, E.; Ha, T.; Deschênes, I. Voltage-Gated Sodium Channels Assemble and Gate as Dimers. Nat. Commun. 2017, 8, 2077. [Google Scholar] [CrossRef]
- Clatot, J.; Zheng, Y.; Girardeau, A.; Liu, H.; Laurita, K.R.; Marionneau, C.; Deschênes, I. Mutant Voltage-Gated Na+ Channels Can Exert a Dominant Negative Effect through Coupled Gating. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1250–H1257. [Google Scholar] [CrossRef]
- Selimi, Z.; Rougier, J.-S.; Abriel, H.; Kucera, J.P. A Detailed Analysis of Single-Channel Nav1.5 Recordings Does Not Reveal Any Cooperative Gating. J. Physiol. 2023, 601, 3847–3868. [Google Scholar] [CrossRef]
- Smits, J.P.P.; Koopmann, T.T.; Wilders, R.; Veldkamp, M.W.; Opthof, T.; Bhuiyan, Z.A.; Mannens, M.M.A.M.; Balser, J.R.; Tan, H.L.; Bezzina, C.R.; et al. A Mutation in the Human Cardiac Sodium Channel (E161K) Contributes to Sick Sinus Syndrome, Conduction Disease and Brugada Syndrome in Two Families. J. Mol. Cell. Cardiol. 2005, 38, 969–981. [Google Scholar] [CrossRef]
- Le Scouarnec, S.; Karakachoff, M.; Gourraud, J.-B.; Lindenbaum, P.; Bonnaud, S.; Portero, V.; Duboscq-Bidot, L.; Daumy, X.; Simonet, F.; Teusan, R.; et al. Testing the Burden of Rare Variation in Arrhythmia-Susceptibility Genes Provides New Insights into Molecular Diagnosis for Brugada Syndrome. Hum. Mol. Genet. 2015, 24, 2757–2763. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Muhammad, A.; Li, B.; Wada, Y.; Hall, L.; Solus, J.F.; Short, L.; Roden, D.M.; Glazer, A.M. Dominant Negative Effects of SCN5A Missense Variants. Genet. Med. 2022, 24, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kimoto, H.; Mishima, H.; Yamagata, K.; Ogata, S.; Aizawa, Y.; Hayashi, K.; Morita, H.; Nakajima, T.; Nakano, Y.; et al. Functionally Validated SCN5A Variants Allow Interpretation of Pathogenicity and Prediction of Lethal Events in Brugada Syndrome. Eur. Heart J. 2021, 42, 2854–2863. [Google Scholar] [CrossRef] [PubMed]
- Pearman, C.M.; Denham, N.C.; Mills, R.W.; Ding, W.Y.; Modi, S.S.; Hall, M.C.S.; Todd, D.M.; Mahida, S. Relationship between Sodium Channel Function and Clinical Phenotype in SCN5A Variants Associated with Brugada Syndrome. Hum. Mutat. 2020, 41, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Crotti, L.; George, A.L. Modifier Genes for Sudden Cardiac Death. Eur. Heart J. 2018, 39, 3925–3931. [Google Scholar] [CrossRef] [PubMed]
- Kroncke, B.M.; Smith, D.K.; Zuo, Y.; Glazer, A.M.; Roden, D.M.; Blume, J.D. A Bayesian Method to Estimate Variant-Induced Disease Penetrance. PLoS Genet. 2020, 16, e1008862. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Pan, X.; Wang, Z.; Yi, M.; Xie, S.; Zhang, X.; Tao, D.; Yang, Y.; Liu, Y. SCN5A-L256del and L1621F Exhibit Loss-of-Function Properties Related to Autosomal Recessive Congenital Cardiac Disorders Presenting as Sick Sinus Syndrome, Dilated Cardiomyopathy, and Sudden Cardiac Death. Gene 2024, 898, 148093. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Rook, M.B.; Groenewegen, W.A.; Herfst, L.J.; van der Wal, A.C.; Lam, J.; Jongsma, H.J.; Wilde, A.A.M.; Mannens, M.M.A.M. Compound Heterozygosity for Mutations (W156X and R225W) in SCN5A Associated with Severe Cardiac Conduction Disturbances and Degenerative Changes in the Conduction System. Circ. Res. 2003, 92, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Caballero, R.; Utrilla, R.G.; Amorós, I.; Matamoros, M.; Pérez-Hernández, M.; Tinaquero, D.; Alfayate, S.; Nieto-Marín, P.; Guerrero-Serna, G.; Liu, Q.-H.; et al. Tbx20 Controls the Expression of the KCNH2 Gene and of hERG Channels. Proc. Natl. Acad. Sci. USA 2017, 114, E416–E425. [Google Scholar] [CrossRef]
- Nieto-Marín, P.; Tinaquero, D.; Utrilla, R.G.; Cebrián, J.; González-Guerra, A.; Crespo-García, T.; Cámara-Checa, A.; Rubio-Alarcón, M.; Dago, M.; Alfayate, S.; et al. Tbx5 Variants Disrupt Nav1.5 Function Differently in Patients Diagnosed with Brugada or Long QT Syndrome. Cardiovasc. Res. 2022, 118, 1046–1060. [Google Scholar] [CrossRef]
- Cámara-Checa, A.; Perin, F.; Rubio-Alarcón, M.; Dago, M.; Crespo-García, T.; Rapún, J.; Marín, M.; Cebrián, J.; Gómez, R.; Bermúdez-Jiménez, F.; et al. A Gain-of-Function HCN4 Mutant in the HCN Domain Is Responsible for Inappropriate Sinus Tachycardia in a Spanish Family. Proc. Natl. Acad. Sci. USA 2023, 120, e2305135120. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; Matamoros, M.; Alfayate, S.; Nieto-Marín, P.; Utrilla, R.G.; Tinaquero, D.; de Andrés, R.; Crespo, T.; Ponce-Balbuena, D.; Willis, B.C.; et al. Brugada Syndrome Trafficking-Defective Nav1.5 Channels Can Trap Cardiac Kir2.1/2.2 Channels. JCI Insight 2018, 3, 96291. [Google Scholar] [CrossRef] [PubMed]
- Dago, M.; Crespo-García, T.; Cámara-Checa, A.; Rapún, J.; Rubio-Alarcón, M.; Marín, M.; Tamargo, J.; Caballero, R.; Delpón, E. Empagliflozin and Dapagliflozin Increase Na+ and Inward Rectifier K+ Current Densities in Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs). Cells 2022, 11, 3707. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Marín, P.; Jiménez-Jáimez, J.; Tinaquero, D.; Alfayate, S.; Utrilla, R.G.; Rodríguez Vázquez Del Rey, M.D.M.; Perin, F.; Sarquella-Brugada, G.; Monserrat, L.; Brugada, J.; et al. Digenic Heterozigosity in SCN5A and CACNA1C Explains the Variable Expressivity of the Long QT Phenotype in a Spanish Family. Rev. Esp. Cardiol. (Engl. Ed.) 2019, 72, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Akai, J.; Makita, N.; Sakurada, H.; Shirai, N.; Ueda, K.; Kitabatake, A.; Nakazawa, K.; Kimura, A.; Hiraoka, M. A Novel SCN5A Mutation Associated with Idiopathic Ventricular Fibrillation without Typical ECG Findings of Brugada Syndrome. FEBS Lett. 2000, 479, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, D.W.; Alings, M.; Fish, F.; Wathen, M.; Roden, D.M.; George, A.L. Congenital Long-QT Syndrome Caused by a Novel Mutation in a Conserved AcidicDomain of the Cardiac Na+ Channel. Circulation 1999, 99, 3165–3171. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Suresh, S.; Gopi, S.; Raman, K.; Naganathan, A.N. A General Mechanism for the Propagation of Mutational Effects in Proteins. Biochemistry 2017, 56, 294–305. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Delgado, M.; Cámara-Checa, A.; Rubio-Alarcón, M.; Heredero-Jung, D.; de la Fuente-Blanco, L.; Rapún, J.; Plata-Izquierdo, B.; Pérez-Martín, S.; Cebrián, J.; Moreno de Redrojo, L.; et al. Variable Penetrance and Expressivity of a Rare Pore Loss-of-Function Mutation (p.L889V) of Nav1.5 Channels in Three Spanish Families. Int. J. Mol. Sci. 2024, 25, 4686. https://doi.org/10.3390/ijms25094686
Gallego-Delgado M, Cámara-Checa A, Rubio-Alarcón M, Heredero-Jung D, de la Fuente-Blanco L, Rapún J, Plata-Izquierdo B, Pérez-Martín S, Cebrián J, Moreno de Redrojo L, et al. Variable Penetrance and Expressivity of a Rare Pore Loss-of-Function Mutation (p.L889V) of Nav1.5 Channels in Three Spanish Families. International Journal of Molecular Sciences. 2024; 25(9):4686. https://doi.org/10.3390/ijms25094686
Chicago/Turabian StyleGallego-Delgado, María, Anabel Cámara-Checa, Marcos Rubio-Alarcón, David Heredero-Jung, Laura de la Fuente-Blanco, Josu Rapún, Beatriz Plata-Izquierdo, Sara Pérez-Martín, Jorge Cebrián, Lucía Moreno de Redrojo, and et al. 2024. "Variable Penetrance and Expressivity of a Rare Pore Loss-of-Function Mutation (p.L889V) of Nav1.5 Channels in Three Spanish Families" International Journal of Molecular Sciences 25, no. 9: 4686. https://doi.org/10.3390/ijms25094686
APA StyleGallego-Delgado, M., Cámara-Checa, A., Rubio-Alarcón, M., Heredero-Jung, D., de la Fuente-Blanco, L., Rapún, J., Plata-Izquierdo, B., Pérez-Martín, S., Cebrián, J., Moreno de Redrojo, L., García-Berrocal, B., Delpón, E., Sánchez, P. L., Villacorta, E., & Caballero, R. (2024). Variable Penetrance and Expressivity of a Rare Pore Loss-of-Function Mutation (p.L889V) of Nav1.5 Channels in Three Spanish Families. International Journal of Molecular Sciences, 25(9), 4686. https://doi.org/10.3390/ijms25094686