
Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: From Bedside to Bench and Back Again

Abstract
:1. Introduction
2. Clinical Aspects and Definitions
2.1. Systemic Sclerosis
2.2. Pulmonary Hypertension—WHO Classification
2.3. SSc PH and SSc-PAH
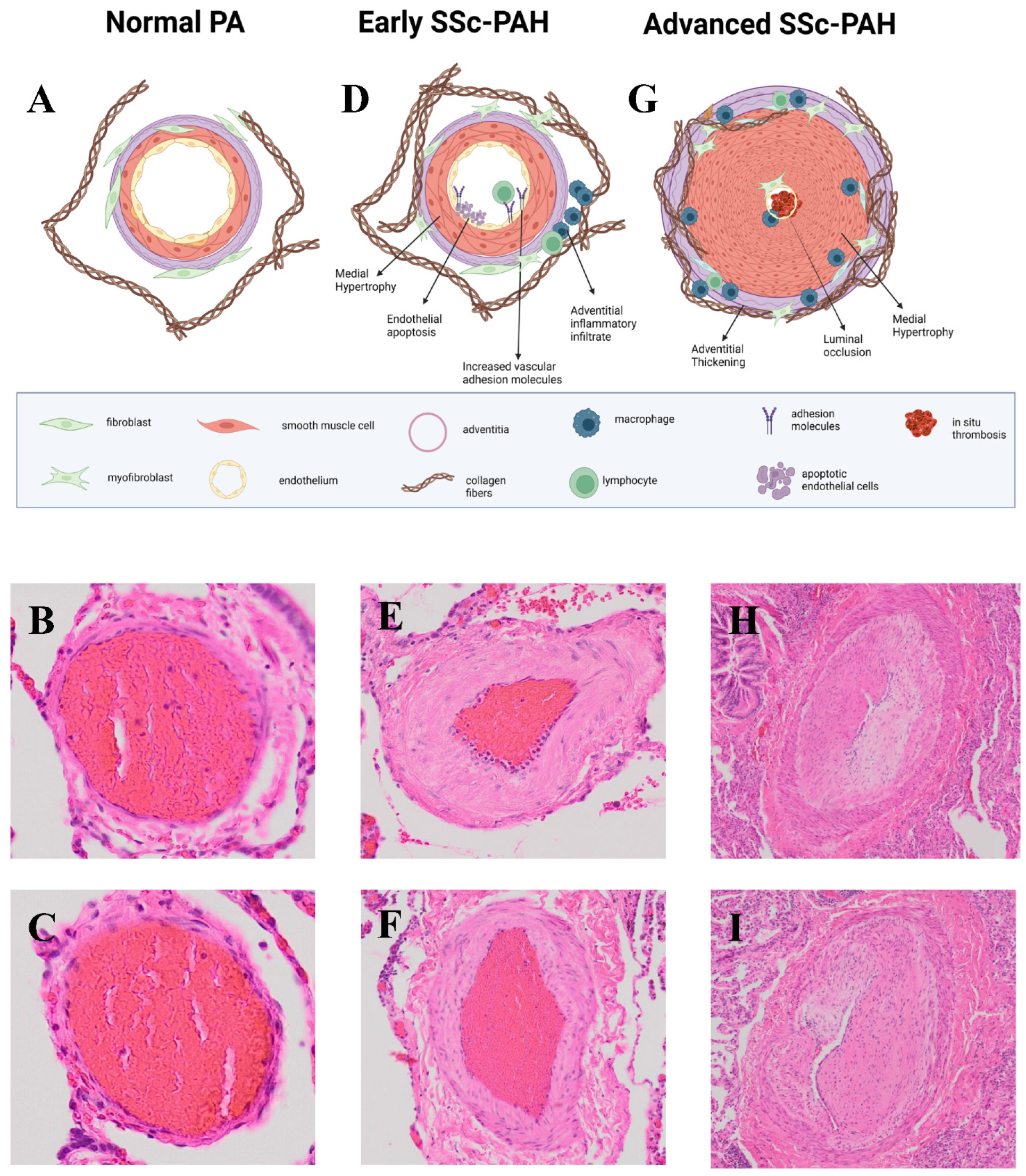
3. Pathology of PAH
SSc-PAH vs. IPAH
4. PAH Genetics
5. Biomarkers
5.1. NT-proBNP and BNP
5.2. Autoantibodies
5.3. Proteome-Wide SSc-PAH Biomarkers
5.4. Metabolic Biomarkers
5.5. Cytokines and Chemokines
5.6. Additional Candidate Biomarkers

{kind=link}
{kind=link}
| Biomarker(s) | Comparison Groups | Association(s) | Reference | |
|---|---|---|---|---|
| Natriuretic Peptides | NT-proBNP | SSc-AR-PAH | Pulmonary Hypertension Severity (mPaP, PVR, Cardiac output, 6MWD, NYHA functional class) | [39] |
| BNP | SSc-AR-PAH | Predictors of progression to SSc-PAH from SSc-AR-PAH (BNP: HR (95% CI) 0.6 (0.1–5.7); NT-proBNP: 1.6 (0.2–14.3), composite BNP/NT-proBNP group predicted mortality (HR 3.81 (2.08–6.99), p < 0.0001) | [39] | |
| Autoantibodies | Anti-centromere, Anti-U3 RNP, Anti-Th/To, Antiphospholipid | SSc with alterative antibodies | SSc-PAH incidence | [40] |
| Anti-ANPA32a | Anti-ANPA32 negative SSc | Echocardiographic evidence of pulmonary hypertension (69% versus 37%; p = 0.012) | [42] | |
| Antibodies against Endothelin 1 | SSc no-PAH, IPAH | Active SSc-PAH (SSc-PAH vs. IPAH: ATR1: 68.8/85.5 (0.772) Anti-ETAR: 72.5/85.5 (0.786)) (Non–SSc-PAH vs. SSc-PAH: ATR1: 68.8/78.0 (0.735) Anti-ETAR: 70.0/82.4 (0.754)) | [43] | |
| Ang receptor type 1 (AT1R) | SSc no-PAH, IPAH | Mortality (anti-AT1R: 68.2% and a specificity of 62.2% (AUC = 0.669; p = 0.03) and Anti-ETAR antibodies: sensitivity of 68.2% and a specificity of 71.1% (AUC = 0.672; p = 0.02). | [43] | |
| Proteome-wide SSc-PAH Biomarkers | RAGE, MMP2, collagen IV, endostatin, neurolipin-1, IGFBP-2, NT-proBNP, IGFBP7 | SSc no-PH | AUC 0.741, sensitivity of 65.2% and a specificity of 68.9% | [45] |
| Chemerin | SSc-no-PAH, HC | Correlates with PVR (ρ = 0.42, p = 0.04) | [46] | |
| Metabolic biomarkers | Nervonic acid, Lignoceric acid, Eicosanoids/oxylipins, Sex hormone metabolites | IPAH, SSc no-PH, SSc-PH | Present in SSc-PAH not in IPAH (85.5% of accuracy (95% CI, 82.8–88.3) | [48] |
| Kynurenine, kynurenine to tryphophan ratio | pre-SSc-PAH, SSc no-PAH | Precursor to SSc-PAH, severity of disease, shorter survival times | [49] | |
| Cytokines | CXCL4 | SSc no-PAH | Precursor to SSc-PAH, earlier development of pulmonary arterial hypertension as determined on right-heart catheterization (HR 8.33; 95% CI, 4.43 to 15.72; p < 0.001) | [50] |
| CCL21 | iPAH, SSc-PAH, SSc non-PAH, HC | Mortality (HR 2.1, 95% CI 1.21–3.70 [p = 0.008]) | [51] | |
| IL-32 | SSc non-PAH, iPAH, HC | mPAP and sPAP levels | [52] | |
| PAI-1, sICAM-1, BDNF, VEGF-D | SSc-High risk for PAH, SSc-Low risk PAH, HC | Profile for patients at high risk for SSc-PAH based on right heart catheterization | [53] | |
| Additional Candidate Biomarkers | Lysyl oxidase (LOX) | Later-SSc, Early-SSc, PRP, HC | Inversely correlated with DLCO | [54] |
| Pentraxin (PTX-3) | healthy controls, SSc-PH, SSc-high risk for PH, SSc-low risk PH | High risk for SSc-PAH (High risk: diffusion capacity (DLco) less than 55% with a forced vital capacity (FVC) greater than 70%, an FVC/Dlco ratio of >1.6, or a right ventricular systolic pressure on an echocardiogram greater than or equal to 40 mm Hg) | [55] | |
| Soluble fms-like tyrosine kinase 1 (sFlt-1), Placenta growth factor (PlGF) | SSc-PH, SSc-no PH | sFlt-1 (p = 0.3245; p = 0.01) positively correlated with right ventricular systolic pressure. PlGF (p = 0.03) and sFlt-1 (p = 0.04) positively correlated with the ratio of forced vital capacity to diffusing capacity for carbon monoxide (DLCO), and both inversely correlated with DLCO (p = 0.01) | [56] | |
| Micro-RNAs: miR-20a-5p and miR-203a-3p | lcSSc-ACA, SSc-APAH, SSc-no PAH | Occurrence of SSc-APAH in female patients with ACA-positive lcSSc | [57] |
6. PAH Animal Models
- A.
- Models of IPAH
6.1. Monocrotaline
6.2. Sugen/Hypoxia
6.3. BMPR2 Transgenic/Knockout Animals
- B.
- Models of Scleroderma or Connective Tissue Disease with Pulmonary Vasculopathy
6.4. Fra-2 Transgenic Mice
6.5. Fli-1/Klf5 Mice
6.6. TNF Transgenic Mice
7. Cellular Pathogenesis
7.1. Endothelial Cells
7.2. Vascular Smooth Muscle Cell
7.3. Fibroblasts
7.4. Pericytes
7.5. Myeloid Cells
7.6. Lymphocytes
8. Intracellular Signaling
8.1. BMP Signaling Pathway
8.2. TGF-β Signaling Pathways
8.3. Vasodilatory Pathways
8.3.1. Nitric Oxide Pathway
8.3.2. Endothelin Pathway
8.3.3. Prostacyclin Pathway
8.4. Notch Pathway
8.5. HIF Pathway
9. Drug Therapy of PAH and SSc-PAH
9.1. Endothelin Receptor Antagonists
9.2. Nitric Oxide: Phosphodiesterase Inhibitors and Guanylate Cyclase Agonists
9.3. Prostacyclin Agonists and Receptor Agonists
9.4. Immunomodulation
9.5. Iron Supplementation and Anticoagulation
9.6. Sotatercept
9.7. Combination Therapy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tuhy, T.; Hassoun, P.M. Clinical features of pulmonary arterial hypertension associated with systemic sclerosis. Front. Med. 2023, 10, 1264906. [Google Scholar] [CrossRef] [PubMed]
- Bairkdar, M.; Rossides, M.; Westerlind, H.; Hesselstrand, R.; Arkema, E.V.; Holmqvist, M. Incidence and prevalence of systemic sclerosis globally: A comprehensive systematic review and meta-analysis. Rheumatology 2021, 60, 3121–3133. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Peacock, A.J.; Murphy, N.F.; McMurray, J.J.; Caballero, L.; Stewart, S. An epidemiological study of pulmonary arterial hypertension. Eur. Respir. J. 2007, 30, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Airo, P.; Meune, C.; Beretta, L.; Dieude, P.; Caramaschi, P.; Tiev, K.; Cappelli, S.; Diot, E.; Vacca, A.; et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J. Rheumatol. 2010, 37, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.J.; Naranjo, M.; Ayoub, N.; Housten, T.; Hsu, S.; Balasubramanian, A.; Simpson, C.E.; Damico, R.L.; Mathai, S.C.; Kolb, T.M.; et al. Improved Survival for Patients with Systemic Sclerosis-associated Pulmonary Arterial Hypertension: The Johns Hopkins Registry. Am. J. Respir. Crit. Care Med. 2023, 207, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Kiely, D.G.; Kovacs, G.; Thompson, A.A.R.; Condliffe, R. Pulmonary hypertension phenotypes in patients with systemic sclerosis. Eur. Respir. Rev. 2021, 30, 210053. [Google Scholar] [CrossRef] [PubMed]
- Rose-Jones, L.J.; McLaughlin, V.V. Pulmonary hypertension: Types and treatments. Curr. Cardiol. Rev. 2015, 11, 73–79. [Google Scholar] [CrossRef]
- Fontes Oliveira, M.; Rei, A.L.; Oliveira, M.I.; Almeida, I.; Santos, M. Prevalence and prognostic significance of heart failure with preserved ejection fraction in systemic sclerosis. Future Cardiol. 2022, 18, 17–25. [Google Scholar] [CrossRef]
- Roofeh, D.; Lin, C.J.F.; Goldin, J.; Kim, G.H.; Furst, D.E.; Denton, C.P.; Huang, S.; Khanna, D.; Focu, S.I. Tocilizumab Prevents Progression of Early Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2021, 73, 1301–1310. [Google Scholar] [CrossRef]
- Sangani, R.A.; Lui, J.K.; Gillmeyer, K.R.; Trojanowski, M.A.; Bujor, A.M.; LaValley, M.P.; Klings, E.S. Clinical characteristics and outcomes in pulmonary manifestations of systemic sclerosis: Contribution from pulmonary hypertension and interstitial lung disease severity. Pulm. Circ. 2022, 12, e12117. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Madani, M.M.; Mahmud, E.; Kim, N.H. Evaluation and Management of Chronic Thromboembolic Pulmonary Hypertension. Chest 2023, 164, 490–502. [Google Scholar] [CrossRef]
- Rubio-Rivas, M.; Homs, N.A.; Cuartero, D.; Corbella, X. The prevalence and incidence rate of pulmonary arterial hypertension in systemic sclerosis: Systematic review and meta-analysis. Autoimmun. Rev. 2021, 20, 102713. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.T.; Lee, P.; Gladman, D.D.; Abu-Shakra, M. Pulmonary hypertension in systemic sclerosis: An analysis of 17 patients. Br. J. Rheumatol. 1996, 35, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Zhao, C.; Saggar, R.; Mathai, S.C.; Chung, L.; Coghlan, J.G.; Shah, M.; Hartney, J.; McLaughlin, V. Long-Term Outcomes in Patients with Connective Tissue Disease-Associated Pulmonary Arterial Hypertension in the Modern Treatment Era: Meta-Analyses of Randomized, Controlled Trials and Observational Registries. Arthritis Rheumatol. 2021, 73, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Galambos, C.; Sims-Lucas, S.; Abman, S.H.; Cool, C.D. Intrapulmonary Bronchopulmonary Anastomoses and Plexiform Lesions in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2016, 193, 574–576. [Google Scholar] [CrossRef]
- Farber, H.W.; Loscalzo, J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef]
- Dorfmuller, P.; Montani, D.; Humbert, M. Beyond arterial remodelling: Pulmonary venous and cardiac involvement in patients with systemic sclerosis-associated pulmonary arterial hypertension. Eur. Respir. J. 2010, 35, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, M.J.; Vonk, M.C.; Boonstra, A.; Voskuyl, A.E.; Vonk-Noordegraaf, A.; Smit, E.F.; Dijkmans, B.A.; Postmus, P.E.; Mooi, W.J.; Heijdra, Y.; et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: A distinctive vasculopathy. Eur. Respir. J. 2009, 34, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Dorfmuller, P.; Humbert, M.; Perros, F.; Sanchez, O.; Simonneau, G.; Muller, K.M.; Capron, F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum. Pathol. 2007, 38, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, M.J.; Mouchaers, K.T.; Niessen, H.M.; Hadi, A.M.; Kupreishvili, K.; Boonstra, A.; Voskuyl, A.E.; Belien, J.A.; Smit, E.F.; Dijkmans, B.C.; et al. Characteristics of interstitial fibrosis and inflammatory cell infiltration in right ventricles of systemic sclerosis-associated pulmonary arterial hypertension. Int. J. Rheumatol. 2010, 2010, 604615. [Google Scholar] [CrossRef]
- Tedford, R.J.; Mudd, J.O.; Girgis, R.E.; Mathai, S.C.; Zaiman, A.L.; Housten-Harris, T.; Boyce, D.; Kelemen, B.W.; Bacher, A.C.; Shah, A.A.; et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ. Heart Fail. 2013, 6, 953–963. [Google Scholar] [CrossRef]
- Hsu, S.; Kokkonen-Simon, K.M.; Kirk, J.A.; Kolb, T.M.; Damico, R.L.; Mathai, S.C.; Mukherjee, M.; Shah, A.A.; Wigley, F.M.; Margulies, K.B.; et al. Right Ventricular Myofilament Functional Differences in Humans with Systemic Sclerosis-Associated Versus Idiopathic Pulmonary Arterial Hypertension. Circulation 2018, 137, 2360–2370. [Google Scholar] [CrossRef]
- Ma, F.; Tsou, P.S.; Gharaee-Kermani, M.; Plazyo, O.; Xing, X.; Kirma, J.; Wasikowski, R.; Hile, G.A.; Harms, P.W.; Jiang, Y.; et al. Systems-based identification of the Hippo pathway for promoting fibrotic mesenchymal differentiation in systemic sclerosis. Nat. Commun. 2024, 15, 210. [Google Scholar] [CrossRef]
- Rius Rigau, A.; Li, Y.N.; Matei, A.E.; Gyorfi, A.H.; Bruch, P.M.; Koziel, S.; Devakumar, V.; Gabrielli, A.; Kreuter, A.; Wang, J.; et al. Characterization of Vascular Niche in Systemic Sclerosis by Spatial Proteomics. Circ. Res. 2024, 134, 875–891. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef]
- Boucherat, O.; Agrawal, V.; Lawrie, A.; Bonnet, S. The Latest in Animal Models of Pulmonary Hypertension and Right Ventricular Failure. Circ. Res. 2022, 130, 1466–1486. [Google Scholar] [CrossRef]
- Happe, C.; Kurakula, K.; Sun, X.Q.; da Silva Goncalves Bos, D.; Rol, N.; Guignabert, C.; Tu, L.; Schalij, I.; Wiesmeijer, K.C.; Tura-Ceide, O.; et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Halley, G.R.; Golpon, H.A.; Voelkel, N.F.; Tuder, R.M. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ. Res. 2001, 88, E2–E11. [Google Scholar] [CrossRef] [PubMed]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Zhao, M.; West, J.; Newman, J.H.; Rich, S.; Archer, S.L.; Robbins, I.M.; Blackwell, T.S.; Cogan, J.; Loyd, J.E.; et al. Critical Genomic Networks and Vasoreactive Variants in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 464–475. [Google Scholar] [CrossRef]
- Unlu, B.; Tursen, U.; Rajabi, Z.; Jabalameli, N.; Rajabi, F. The Immunogenetics of Systemic Sclerosis. Adv. Exp. Med. Biol. 2022, 1367, 259–298. [Google Scholar] [PubMed]
- Koumakis, E.; Wipff, J.; Dieude, P.; Ruiz, B.; Bouaziz, M.; Revillod, L.; Guedj, M.; Distler, J.H.; Matucci-Cerinic, M.; Humbert, M.; et al. TGFbeta receptor gene variants in systemic sclerosis-related pulmonary arterial hypertension: Results from a multicentre EUSTAR study of European Caucasian patients. Ann. Rheum. Dis. 2012, 71, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Turk, M.A.; Pope, J.E. Factors associated with pulmonary arterial hypertension (PAH) in systemic sclerosis (SSc). Autoimmun. Rev. 2020, 19, 102602. [Google Scholar] [CrossRef] [PubMed]
- Hickey, P.M.; Lawrie, A.; Condliffe, R. Circulating Protein Biomarkers in Systemic Sclerosis Related Pulmonary Arterial Hypertension: A Review of Published Data. Front. Med. 2018, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Fairchild, R.M.; Furst, D.E.; Li, S.; Alkassab, F.; Bolster, M.B.; Csuka, M.E.; Derk, C.T.; Domsic, R.T.; Fischer, A.; et al. Utility of B-type natriuretic peptides in the assessment of patients with systemic sclerosis-associated pulmonary hypertension in the PHAROS registry. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 106), 106–113. [Google Scholar]
- Nunes, J.P.L.; Cunha, A.C.; Meirinhos, T.; Nunes, A.; Araujo, P.M.; Godinho, A.R.; Vilela, E.M.; Vaz, C. Prevalence of auto-antibodies associated to pulmonary arterial hypertension in scleroderma—A review. Autoimmun. Rev. 2018, 17, 1186–1201. [Google Scholar] [CrossRef]
- Liem, S.I.E.; Boonstra, M.; Le Cessie, S.; Riccardi, A.; Airo, P.; Distler, O.; Matucci-Cerinic, M.; Caimmi, C.; Siegert, E.; Allanore, Y.; et al. Sex-specific risk of anti-topoisomerase antibodies on mortality and disease severity in systemic sclerosis: 10-year analysis of the Leiden CCISS and EUSTAR cohorts. Lancet Rheumatol. 2022, 4, e699–e709. [Google Scholar] [CrossRef]
- Wallwork, R.; Casciola-Rosen, L.; Shah, A.A. Anti-ANP32A antibodies in systemic sclerosis. Ann. Rheum. Dis. 2022, 81, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kuhl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Hegner, B.; Kretzschmar, T.; Zhu, N.; Kleinau, G.; Zhao, H.; Kamhieh-Milz, J.; Hilger, J.; Schindler, R.; Scheerer, P.; Riemekasten, G.; et al. Autoimmune activation and hypersensitization of the AT1 and ETA receptors contributes to vascular injury in scleroderma renal crisis. Rheumatology 2023, 62, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Bauer, Y.; de Bernard, S.; Hickey, P.; Ballard, K.; Cruz, J.; Cornelisse, P.; Chadha-Boreham, H.; Distler, O.; Rosenberg, D.; Doelberg, M.; et al. Identifying early pulmonary arterial hypertension biomarkers in systemic sclerosis: Machine learning on proteomics from the DETECT cohort. Eur. Respir. J. 2021, 57, 2002591. [Google Scholar] [CrossRef] [PubMed]
- Sanges, S.; Rice, L.; Tu, L.; Valenzi, E.; Cracowski, J.L.; Montani, D.; Mantero, J.C.; Ternynck, C.; Marot, G.; Bujor, A.M.; et al. Biomarkers of haemodynamic severity of systemic sclerosis-associated pulmonary arterial hypertension by serum proteome analysis. Ann. Rheum. Dis. 2023, 82, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.M.; Mantero, J.C.; Stratton, E.A.; Warburton, R.; Roberts, K.; Hill, N.; Simms, R.W.; Domsic, R.; Farber, H.W.; Layfatis, R. Serum biomarker for diagnostic evaluation of pulmonary arterial hypertension in systemic sclerosis. Arthritis Res. Ther. 2018, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.; Shao, J.; Pauciulo, M.W.; Nichols, W.C.; Hemnes, A.R.; Malhotra, A.; Kim, N.H.; Yuan, J.X.; Fernandes, T.; Kerr, K.M.; et al. Metabolomic Profiles Differentiate Scleroderma-PAH from Idiopathic PAH and Correspond with Worsened Functional Capacity. Chest 2023, 163, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.E.; Ambade, A.S.; Harlan, R.; Roux, A.; Aja, S.; Graham, D.; Shah, A.A.; Hummers, L.K.; Hemnes, A.R.; Leopold, J.A.; et al. Kynurenine pathway metabolism evolves with development of preclinical and scleroderma-associated pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2023, 325, L617–L627. [Google Scholar] [CrossRef]
- van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.M.; Hesselstrand, R.; Fretheim, H.; Ueland, T.; Andreassen, A.K.; Brunborg, C.; Palchevskiy, V.; Midtvedt, O.; Garen, T.; Aukrust, P.; et al. CCL21 as a Potential Serum Biomarker for Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis Rheumatol. 2018, 70, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, P.; Guggino, G.; Manzi, G.; Ruscitti, P.; Berardicurti, O.; Panzera, N.; Grazia, N.; Badagliacca, R.; Riccieri, V.; Vizza, C.D.; et al. Interleukin-32 in systemic sclerosis, a potential new biomarker for pulmonary arterial hypertension. Arthritis Res. Ther. 2020, 22, 127. [Google Scholar] [CrossRef] [PubMed]
- Kolstad, K.D.; Khatri, A.; Donato, M.; Chang, S.E.; Li, S.; Steen, V.D.; Utz, P.J.; Khatri, P.; Chung, L. Cytokine signatures differentiate systemic sclerosis patients at high versus low risk for pulmonary arterial hypertension. Arthritis Res. Ther. 2022, 24, 39. [Google Scholar] [CrossRef]
- Vadasz, Z.; Balbir Gurman, A.; Meroni, P.; Farge, D.; Levi, Y.; Ingegnoli, F.; Braun-Moscovici, Y.; Rosner, I.; Slobodin, G.; Rozenbaum, M.; et al. Lysyl oxidase-a possible role in systemic sclerosis-associated pulmonary hypertension: A multicentre study. Rheumatology 2019, 58, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Lammi, M.R.; Kolstad, K.D.; Saketkoo, L.A.; Khatri, A.; Utz, P.J.; Steen, V.D.; Chung, L. Endothelial Biomarkers of Systemic Sclerosis-Associated Pulmonary Hypertension. Arthritis Care Res. 2023, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McMahan, Z.; Schoenhoff, F.; Van Eyk, J.E.; Wigley, F.M.; Hummers, L.K. Biomarkers of pulmonary hypertension in patients with scleroderma: A case-control study. Arthritis Res. Ther. 2015, 17, 201. [Google Scholar] [CrossRef] [PubMed]
- Wuttge, D.M.; Carlsen, A.L.; Teku, G.; Wildt, M.; Radegran, G.; Vihinen, M.; Heegaard, N.H.H.; Hesselstrand, R. Circulating plasma microRNAs in systemic sclerosis-associated pulmonary arterial hypertension. Rheumatology 2021, 61, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Dignam, J.P.; Scott, T.E.; Kemp-Harper, B.K.; Hobbs, A.J. Animal models of pulmonary hypertension: Getting to the heart of the problem. Br. J. Pharmacol. 2022, 179, 811–837. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.S.; Gillespie, M.N.; McMurtry, I.F. Fifty Years of Monocrotaline-Induced Pulmonary Hypertension: What Has It Meant to the Field? Chest 2017, 152, 1106–1108. [Google Scholar] [CrossRef]
- Toba, M.; Alzoubi, A.; O’Neill, K.D.; Gairhe, S.; Matsumoto, Y.; Oshima, K.; Abe, K.; Oka, M.; McMurtry, I.F. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H243–H250. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Sung, Y.K.; Shuffle, E.; Wu, T.H.; Kao, P.N.; Tu, A.B.; Dorfmuller, P.; Cao, A.; Wang, L.; et al. Phenotypically Silent Bone Morphogenetic Protein Receptor 2 Mutations Predispose Rats to Inflammation-Induced Pulmonary Arterial Hypertension by Enhancing the Risk for Neointimal Transformation. Circulation 2019, 140, 1409–1425. [Google Scholar] [CrossRef]
- Yue, X.; Yu, X.; Petersen, F.; Riemekasten, G. Recent advances in mouse models for systemic sclerosis. Autoimmun. Rev. 2018, 17, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Hasselblatt, P.; Rath, M.; Popper, H.; Zenz, R.; Komnenovic, V.; Idarraga, M.H.; Kenner, L.; Wagner, E.F. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc. Natl. Acad. Sci. USA 2008, 105, 10525–10530. [Google Scholar] [CrossRef]
- Bell, R.D.; White, R.J.; Garcia-Hernandez, M.L.; Wu, E.; Rahimi, H.; Marangoni, R.G.; Slattery, P.; Duemmel, S.; Nuzzo, M.; Huertas, N.; et al. Tumor Necrosis Factor Induces Obliterative Pulmonary Vascular Disease in a Novel Model of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2020, 72, 1759–1770. [Google Scholar] [CrossRef]
- Wu, X.H.; Ma, J.L.; Ding, D.; Ma, Y.J.; Wei, Y.P.; Jing, Z.C. Experimental animal models of pulmonary hypertension: Development and challenges. Anim. Models Exp. Med. 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Ruiter, G.; de Man, F.S.; Schalij, I.; Sairras, S.; Grunberg, K.; Westerhof, N.; van der Laarse, W.J.; Vonk-Noordegraaf, A. Reversibility of the monocrotaline pulmonary hypertension rat model. Eur. Respir. J. 2013, 42, 553–556. [Google Scholar] [CrossRef]
- Bueno-Beti, C.; Sassi, Y.; Hajjar, R.J.; Hadri, L. Pulmonary Artery Hypertension Model in Rats by Monocrotaline Administration. Methods Mol. Biol. 2018, 1816, 233–241. [Google Scholar] [PubMed]
- Wilson, D.W.; Segall, H.J.; Pan, L.C.; Dunston, S.K. Progressive inflammatory and structural changes in the pulmonary vasculature of monocrotaline-treated rats. Microvasc. Res. 1989, 38, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Arneson, D.; Umar, S.; Ruffenach, G.; Cunningham, C.M.; Ahn, I.S.; Diamante, G.; Bhetraratana, M.; Park, J.F.; Said, E.; et al. Single-Cell Study of Two Rat Models of Pulmonary Arterial Hypertension Reveals Connections to Human Pathobiology and Drug Repositioning. Am. J. Respir. Crit. Care Med. 2021, 203, 1006–1022. [Google Scholar] [CrossRef]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.G.; Fargnoli, A.S.; Gubara, S.M.; Bisserier, M.; Sassi, Y.; Bridges, C.R.; Hajjar, R.J.; Hadri, L. The Left Pneumonectomy Combined with Monocrotaline or Sugen as a Model of Pulmonary Hypertension in Rats. J. Vis. Exp. 2019, e59050. [Google Scholar]
- Vitali, S.H.; Hansmann, G.; Rose, C.; Fernandez-Gonzalez, A.; Scheid, A.; Mitsialis, S.A.; Kourembanas, S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: Long-term follow-up. Pulm. Circ. 2014, 4, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Moonen, J.R.; Cao, A.; Isobe, S.; Li, C.G.; Tojais, N.F.; Taylor, S.; Marciano, D.P.; Chen, P.I.; Gu, M.; et al. Dysregulated Smooth Muscle Cell BMPR2-ARRB2 Axis Causes Pulmonary Hypertension. Circ. Res. 2023, 132, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Distler, J.H.; Distler, O. The Fra-2 transgenic mouse model of systemic sclerosis. Vasc. Pharmacol. 2013, 58, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Reich, N.; Juengel, A.; Kriegsmann, J.; Gay, R.E.; Schett, G.; Michel, B.A.; Gay, S.; Distler, J.H.; Distler, O. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Looney, A.P.; Han, R.; Stawski, L.; Marden, G.; Iwamoto, M.; Trojanowska, M. Synergistic Role of Endothelial ERG and FLI1 in Mediating Pulmonary Vascular Homeostasis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 121–131. [Google Scholar] [CrossRef]
- Asano, Y.; Markiewicz, M.; Kubo, M.; Szalai, G.; Watson, D.K.; Trojanowska, M. Transcription factor Fli1 regulates collagen fibrillogenesis in mouse skin. Mol. Cell. Biol. 2009, 29, 425–434. [Google Scholar] [CrossRef]
- Asano, Y.; Stawski, L.; Hant, F.; Highland, K.; Silver, R.; Szalai, G.; Watson, D.K.; Trojanowska, M. Endothelial Fli1 deficiency impairs vascular homeostasis: A role in scleroderma vasculopathy. Am. J. Pathol. 2010, 176, 1983–1998. [Google Scholar] [CrossRef]
- Noda, S.; Asano, Y.; Nishimura, S.; Taniguchi, T.; Fujiu, K.; Manabe, I.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Akamata, K.; et al. Simultaneous downregulation of KLF5 and Fli1 is a key feature underlying systemic sclerosis. Nat. Commun. 2014, 5, 5797. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Wu, E.K.; Rudmann, C.A.; Forney, M.; Kaiser, C.R.W.; Wood, R.W.; Chakkalakal, J.V.; Paris, N.D.; Klose, A.; Xiao, G.Q.; et al. Selective Sexual Dimorphisms in Musculoskeletal and Cardiopulmonary Pathologic Manifestations and Mortality Incidence in the Tumor Necrosis Factor-Transgenic Mouse Model of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Duemmel, S.; Nuzzo, M.; Bhattacharya, S.; Xu, Q.; Mohan, A.; Korman, B. TNF Receptor 1 Drives Murine Pulmonary Arterial Hypertension and Is Characterized by Loss of Capillary Endothelial Cells and Pericytes, Smooth Muscle Cell Proliferation, and Alterations in Fibroblast Phenotype. Arthritis Rheumatol. 2022, 74, 3222–3225. [Google Scholar]
- Rangel-Moreno, J.; Garcia-Hernandez, M.; Xu, Q.; Korman, B. TNF-mediated Pulmonary Hypertension Is Marked by Aberrant Bone Morphogenic Protein (BMP) and Integrin/Basement Membrane Ligand-Receptor Signaling. Arthritis Rheumatol. 2023, 75. [Google Scholar]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957. [Google Scholar] [CrossRef]
- Vattulainen-Collanus, S.; Akinrinade, O.; Li, M.; Koskenvuo, M.; Li, C.G.; Rao, S.P.; de Jesus Perez, V.; Yuan, K.; Sawada, H.; Koskenvuo, J.W.; et al. Loss of PPARgamma in endothelial cells leads to impaired angiogenesis. J. Cell Sci. 2016, 129, 693–705. [Google Scholar]
- Le Hiress, M.; Tu, L.; Ricard, N.; Phan, C.; Thuillet, R.; Fadel, E.; Dorfmuller, P.; Montani, D.; de Man, F.; Humbert, M.; et al. Proinflammatory Signature of the Dysfunctional Endothelium in Pulmonary Hypertension. Role of the Macrophage Migration Inhibitory Factor/CD74 Complex. Am. J. Respir. Crit. Care Med. 2015, 192, 983–997. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914. [Google Scholar] [CrossRef]
- Patnaik, E.; Lyons, M.; Tran, K.; Pattanaik, D. Endothelial Dysfunction in Systemic Sclerosis. Int. J. Mol. Sci. 2023, 24, 14385. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Cao, Y.; Qin, J.; Chen, Z.; Hu, G.; Li, Q. Pulmonary artery smooth muscle cell phenotypic switching: A key event in the early stage of pulmonary artery hypertension. Drug Discov. Today 2023, 28, 103559. [Google Scholar] [CrossRef] [PubMed]
- Roostalu, U.; Aldeiri, B.; Albertini, A.; Humphreys, N.; Simonsen-Jackson, M.; Wong, J.K.F.; Cossu, G. Distinct Cellular Mechanisms Underlie Smooth Muscle Turnover in Vascular Development and Repair. Circ. Res. 2018, 122, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Wang, S.Y.; Sheban, F.; Zada, M.; Li, B.; Kharouf, F.; Peleg, H.; Aamar, S.; Yalin, A.; Kirschenbaum, D.; et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell 2022, 185, 1373–1388.e20. [Google Scholar] [CrossRef]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Trejo Bittar, H.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2019, 78, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Tabib, T.; Huang, M.; Morse, N.; Papazoglou, A.; Behera, R.; Jia, M.; Bulik, M.; Monier, D.E.; Benos, P.V.; Chen, W.; et al. Myofibroblast transcriptome indicates SFRP2(hi) fibroblast progenitors in systemic sclerosis skin. Nat. Commun. 2021, 12, 4384. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.T.; Bignold, R.E.; Wu, X.; Johnson, J.R. Pericytes: The lung-forgotten cell type. Front. Physiol. 2023, 14, 1150028. [Google Scholar] [CrossRef] [PubMed]
- Bordenave, J.; Tu, L.; Berrebeh, N.; Thuillet, R.; Cumont, A.; Le Vely, B.; Fadel, E.; Nadaud, S.; Savale, L.; Humbert, M.; et al. Lineage Tracing Reveals the Dynamic Contribution of Pericytes to the Blood Vessel Remodeling in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 766–782. [Google Scholar] [CrossRef]
- Abid, S.; Marcos, E.; Parpaleix, A.; Amsellem, V.; Breau, M.; Houssaini, A.; Vienney, N.; Lefevre, M.; Derumeaux, G.; Evans, S.; et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur. Respir. J. 2019, 54, 1802308. [Google Scholar] [CrossRef]
- Zawia, A.; Arnold, N.D.; West, L.; Pickworth, J.A.; Turton, H.; Iremonger, J.; Braithwaite, A.T.; Canedo, J.; Johnston, S.A.; Thompson, A.A.R.; et al. Altered Macrophage Polarization Induces Experimental Pulmonary Hypertension and Is Observed in Patients with Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q.; Wang, C.C.; Pang, X.B.; Shi, J.Z.; Li, H.R.; Xie, X.M.; Wang, Z.; Zhang, H.D.; Zhou, Y.F.; Chen, J.W.; et al. Role of macrophages in pulmonary arterial hypertension. Front. Immunol. 2023, 14, 1152881. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Dorfmuller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Herve, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Ball, M.S.; Martyanov, V.; Popovich, D.; Schaafsma, E.; Han, S.; ElTanbouly, M.; Orzechowski, N.M.; Carns, M.; Arroyo, E.; et al. Profibrotic Activation of Human Macrophages in Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Makinde, H.M.; Dunn, J.L.M.; Gadhvi, G.; Carns, M.; Aren, K.; Chung, A.H.; Muhammad, L.N.; Song, J.; Cuda, C.M.; Dominguez, S.; et al. Three Distinct Transcriptional Profiles of Monocytes Associate with Disease Activity in Scleroderma Patients. Arthritis Rheumatol. 2023, 75, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, S.; Nicolls, M.R.; Taraseviciene, L.; Speich, R.; Voelkel, N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration 2008, 75, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Rock, M.T.; Mosse, C.A.; Vnencak-Jones, C.L.; Yoder, S.M.; Robbins, I.M.; Loyd, J.E.; Meyrick, B.O. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir. Med. 2010, 104, 454–462. [Google Scholar] [CrossRef]
- Sanges, S.; Guerrier, T.; Duhamel, A.; Guilbert, L.; Hauspie, C.; Largy, A.; Balden, M.; Podevin, C.; Lefevre, G.; Jendoubi, M.; et al. Soluble markers of B cell activation suggest a role of B cells in the pathogenesis of systemic sclerosis-associated pulmonary arterial hypertension. Front. Immunol. 2022, 13, 954007. [Google Scholar] [CrossRef]
- Dib, H.; Tamby, M.C.; Bussone, G.; Regent, A.; Berezne, A.; Lafine, C.; Broussard, C.; Simonneau, G.; Guillevin, L.; Witko-Sarsat, V.; et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur. Respir. J. 2012, 39, 1405–1414. [Google Scholar] [CrossRef]
- Tamby, M.C.; Humbert, M.; Guilpain, P.; Servettaz, A.; Dupin, N.; Christner, J.J.; Simonneau, G.; Fermanian, J.; Weill, B.; Guillevin, L.; et al. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur. Respir. J. 2006, 28, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Rol, N.; Kurakula, K.B.; Happe, C.; Bogaard, H.J.; Goumans, M.J. TGF-beta and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. [Google Scholar] [CrossRef]
- Dannewitz Prosseda, S.; Ali, M.K.; Spiekerkoetter, E. Novel Advances in Modifying BMPR2 Signaling in PAH. Genes 2020, 12, 8. [Google Scholar] [CrossRef]
- Gilbane, A.J.; Derrett-Smith, E.; Trinder, S.L.; Good, R.B.; Pearce, A.; Denton, C.P.; Holmes, A.M. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-beta-dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am. J. Respir. Crit. Care Med. 2015, 191, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Jiang, Y.; Li, Q.; Liu, C.; Hu, F.; Li, M. Bone morphogenetic protein-7 inhibits endothelial-to-mesenchymal transition in primary human umbilical vein endothelial cells and mouse model of systemic sclerosis via Akt/mTOR/p70S6K pathway. J. Dermatol. Sci. 2021, 103, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Sheppard, D. TGF-beta signaling in health and disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef]
- Lafyatis, R. Transforming growth factor beta—At the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- Khadilkar, P.; Chougule, D.; Tipnis, T.; Khopkar, U.; Nadkar, M.; Rajadhyaksha, A.; Kini, S.; Kharkar, V.; Athvale, A.; Athvale, T.; et al. A comparative study of modulatory interaction between cytokines and apoptotic proteins among Scleroderma patients with and without pulmonary involvement. Cytokine 2023, 166, 156183. [Google Scholar] [CrossRef]
- Zaaroor Levy, M.; Rabinowicz, N.; Yamila Kohon, M.; Shalom, A.; Berl, A.; Hornik-Lurie, T.; Drucker, L.; Tartakover Matalon, S.; Levy, Y. MiRNAs in Systemic Sclerosis Patients with Pulmonary Arterial Hypertension: Markers and Effectors. Biomedicines 2022, 10, 629. [Google Scholar] [CrossRef]
- Qaiser, K.N.; Tonelli, A.R. Novel Treatment Pathways in Pulmonary Arterial Hypertension. Methodist DeBakey Cardiovasc. J. 2021, 17, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, B.; Xu, H.; Zhang, X. The Emerging Therapeutic Role of Prostaglandin E2 Signaling in Pulmonary Hypertension. Metabolites 2023, 13, 1152. [Google Scholar] [CrossRef] [PubMed]
- Kozij, N.K.; Granton, J.T.; Silkoff, P.E.; Thenganatt, J.; Chakravorty, S.; Johnson, S.R. Exhaled Nitric Oxide in Systemic Sclerosis Lung Disease. Can. Respir. J. 2017, 2017, 6736239. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Kadowitz, P.J. The Nitric Oxide Pathway in Pulmonary Vascular Disease. Am. J. Cardiol. 2017, 120, S71–S79. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Bowman, L.; D’Arsigny, C.L.; Archer, S.L. Soluble guanylate cyclase: A new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clin. Pharmacol. Ther. 2015, 97, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Gupta, M.; Xu, W.; Mavrakis, D.A.; Janocha, A.J.; Comhair, S.A.; Haque, M.M.; Stuehr, D.J.; Yu, J.; Polgar, P.; et al. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1199–L1205. [Google Scholar] [CrossRef]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Kharitonov, S.A.; Cailes, J.B.; Black, C.M.; du Bois, R.M.; Barnes, P.J. Decreased nitric oxide in the exhaled air of patients with systemic sclerosis with pulmonary hypertension. Thorax 1997, 52, 1051–1055. [Google Scholar] [CrossRef]
- Kanai, S.M.; Clouthier, D.E. Endothelin signaling in development. Development 2023, 150, dev201786. [Google Scholar] [CrossRef]
- Rokni, M.; Sadeghi Shaker, M.; Kavosi, H.; Shokoofi, S.; Mahmoudi, M.; Farhadi, E. The role of endothelin and RAS/ERK signaling in immunopathogenesis-related fibrosis in patients with systemic sclerosis: An updated review with therapeutic implications. Arthritis Res. Ther. 2022, 24, 108. [Google Scholar] [CrossRef]
- Morelli, S.; Ferri, C.; Di Francesco, L.; Baldoncini, R.; Carlesimo, M.; Bottoni, U.; Properzi, G.; Santucci, A. Plasma endothelin-1 levels in patients with systemic sclerosis: Influence of pulmonary or systemic arterial hypertension. Ann. Rheum. Dis. 1995, 54, 730–734. [Google Scholar] [CrossRef]
- Hajialilo, M.; Noorabadi, P.; Tahsini Tekantapeh, S.; Malek Mahdavi, A. Endothelin-1, alpha-Klotho, 25(OH) Vit D levels and severity of disease in scleroderma patients. Rheumatol. Int. 2017, 37, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Liu, J.; Zheng, X.; Hu, X.; He, Y. Prostaglandin and prostaglandin receptors: Present and future promising therapeutic targets for pulmonary arterial hypertension. Respir. Res. 2023, 24, 263. [Google Scholar] [CrossRef]
- Cathcart, M.C.; Tamosiuniene, R.; Chen, G.; Neilan, T.G.; Bradford, A.; O’Byrne, K.J.; Fitzgerald, D.J.; Pidgeon, G.P. Cyclooxygenase-2-linked attenuation of hypoxia-induced pulmonary hypertension and intravascular thrombosis. J. Pharmacol. Exp. Ther. 2008, 326, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Falcetti, E.; Hall, S.M.; Phillips, P.G.; Patel, J.; Morrell, N.W.; Haworth, S.G.; Clapp, L.H. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1161–1170. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: Developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc. and the Pulmonary Hypertension Association. Circulation 2009, 119, 2250–2294. [Google Scholar] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef]
- Bodas, M.; Subramaniyan, B.; Karmouty-Quintana, H.; Vitiello, P.F.; Walters, M.S. The emerging role of NOTCH3 receptor signalling in human lung diseases. Expert. Rev. Mol. Med. 2022, 24, e33. [Google Scholar] [CrossRef]
- Zmorzynski, S.; Styk, W.; Filip, A.A.; Krasowska, D. The Significance of NOTCH Pathway in the Development of Fibrosis in Systemic Sclerosis. Ann. Dermatol. 2019, 31, 365–371. [Google Scholar] [CrossRef]
- Xiao, Y.; Gong, D.; Wang, W. Soluble JAGGED1 inhibits pulmonary hypertension by attenuating notch signaling. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2733–2739. [Google Scholar] [CrossRef]
- Miyagawa, K.; Shi, M.; Chen, P.I.; Hennigs, J.K.; Zhao, Z.; Wang, M.; Li, C.G.; Saito, T.; Taylor, S.; Sa, S.; et al. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ. Res. 2019, 124, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Li, Y.; de Jesus, D.; Sembrat, J.; Rojas, M.M.; Goncharova, E.; Cifuentes-Pagano, E.; Straub, A.C.; Pagano, P.J. Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H542–H557. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef]
- Ramadhiani, R.; Ikeda, K.; Miyagawa, K.; Ryanto, G.R.T.; Tamada, N.; Suzuki, Y.; Kirita, Y.; Matoba, S.; Hirata, K.; Emoto, N. Endothelial cell senescence exacerbates pulmonary hypertension by inducing juxtacrine Notch signaling in smooth muscle cells. iScience 2023, 26, 106662. [Google Scholar] [CrossRef]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef]
- Dees, C.; Tomcik, M.; Zerr, P.; Akhmetshina, A.; Horn, A.; Palumbo, K.; Beyer, C.; Zwerina, J.; Distler, O.; Schett, G.; et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, U.; Stenson, E.; Sahu, M.; Thakur, K.K.; Wang, J.; Shah, A.; Mayes, M.; Doumatey, A.; Bentley, A.; Shriner, D.; et al. Functional Variants Increase Notch Signaling and Susceptibility for Systemic Sclerosis. Arthritis Rheumatol. 2022, 74, 2233–2235. [Google Scholar]
- Kavian, N.; Servettaz, A.; Mongaret, C.; Wang, A.; Nicco, C.; Chereau, C.; Grange, P.; Vuiblet, V.; Birembaut, P.; Diebold, M.D.; et al. Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheum. 2010, 62, 3477–3487. [Google Scholar] [CrossRef]
- Dees, C.; Zerr, P.; Tomcik, M.; Beyer, C.; Horn, A.; Akhmetshina, A.; Palumbo, K.; Reich, N.; Zwerina, J.; Sticherling, M.; et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheum. 2011, 63, 1396–1404. [Google Scholar] [CrossRef]
- Yao, Q.; Xing, Y.; Wang, Z.; Liang, J.; Lin, Q.; Huang, M.; Chen, Y.; Lin, B.; Xu, X.; Chen, W. MiR-16-5p suppresses myofibroblast activation in systemic sclerosis by inhibiting NOTCH signaling. Aging 2020, 13, 2640–2654. [Google Scholar] [CrossRef]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, W.; Wang, L.; Chen, S.; Tian, B.; Huang, K.; Corrigan, C.J.; Ying, S.; Wang, W.; Wang, C. IL-33 Initiates Vascular Remodelling in Hypoxic Pulmonary Hypertension by up-Regulating HIF-1alpha and VEGF Expression in Vascular Endothelial Cells. EBioMedicine 2018, 33, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, K.; Zheng, Q.; Zhang, C.; Tang, H.; Babicheva, A.; Jiang, Q.; Li, M.; Chen, Y.; Carr, S.G.; et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L216–L228. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.T.; Teng, X.; Zhang, L.L.; Chen, J.N.; Liu, Z.; Chen, X.H.; Zhao, S.; Yang, S.; Feng, J.; Yan, X.Y. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat. Commun. 2019, 10, 3551. [Google Scholar] [CrossRef]
- Hu, C.J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, M.; Sikora, M.; Stec, A.; Zaremba, M.; Maciejewski, C.; Pawlik, K.; Rudnicka, L. Hypoxia-Inducible Factor-1alpha (HIF-1alpha) as a Biomarker for Changes in Microcirculation in Individuals with Systemic Sclerosis. Dermatol. Ther. 2023, 13, 1549–1560. [Google Scholar] [CrossRef]
- Mao, J.; Liu, J.; Zhou, M.; Wang, G.; Xiong, X.; Deng, Y. Hypoxia-induced interstitial transformation of microvascular endothelial cells by mediating HIF-1alpha/VEGF signaling in systemic sclerosis. PLoS ONE 2022, 17, e0263369. [Google Scholar] [CrossRef]
- He, X.; Shi, Y.; Zeng, Z.; Tang, B.; Xiao, X.; Yu, J.; Zou, P.; Liu, J.; Xiao, Y.; Luo, Y.; et al. Intimate intertwining of the pathogenesis of hypoxia and systemic sclerosis: A transcriptome integration analysis. Front. Immunol. 2022, 13, 929289. [Google Scholar] [CrossRef]
- Takagi, K.; Kawamoto, M.; Higuchi, T.; Tochimoto, A.; Harigai, M.; Kawaguchi, Y. Single nucleotide polymorphisms of the HIF1A gene are associated with susceptibility to pulmonary arterial hypertension in systemic sclerosis and contribute to SSc-PAH disease severity. Int. J. Rheum. Dis. 2020, 23, 674–680. [Google Scholar] [CrossRef]
- Arefiev, K.; Fiorentino, D.F.; Chung, L. Endothelin Receptor Antagonists for the Treatment of Raynaud’s Phenomenon and Digital Ulcers in Systemic Sclerosis. Int. J. Rheumatol. 2011, 2011, 201787. [Google Scholar] [CrossRef]
- Galie, N.; Barbera, J.A.; Frost, A.E.; Ghofrani, H.A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.L.; Grunig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Hassoun, P.M.; Zamanian, R.T.; Damico, R.; Lechtzin, N.; Khair, R.; Kolb, T.M.; Tedford, R.J.; Hulme, O.L.; Housten, T.; Pisanello, C.; et al. Ambrisentan and Tadalafil Up-Front Combination Therapy in Scleroderma-associated Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 1102–1110. [Google Scholar] [CrossRef]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galie, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Matucci-Cerinic, M.; Denton, C.P.; Furst, D.E.; Mayes, M.D.; Hsu, V.M.; Carpentier, P.; Wigley, F.M.; Black, C.M.; Fessler, B.J.; Merkel, P.A.; et al. Bosentan treatment of digital ulcers related to systemic sclerosis: Results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2011, 70, 32–38. [Google Scholar] [CrossRef]
- Nguyen, V.A.; Eisendle, K.; Gruber, I.; Hugl, B.; Reider, D.; Reider, N. Effect of the dual endothelin receptor antagonist bosentan on Raynaud’s phenomenon secondary to systemic sclerosis: A double-blind prospective, randomized, placebo-controlled pilot study. Rheumatology 2010, 49, 583–587. [Google Scholar] [CrossRef]
- Castellvi, I.; Simeon, C.P.; Sarmiento, M.; Casademont, J.; Corominas, H.; Fonollosa, V. Effect of bosentan in pulmonary hypertension development in systemic sclerosis patients with digital ulcers. PLoS ONE 2020, 15, e0243651. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galie, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Merkel, P.A.; Krieg, T.; Le Brun, F.O.; Marr, A.; Papadakis, K.; Pope, J.; Matucci-Cerinic, M.; Furst, D.E.; et al. Effect of Macitentan on the Development of New Ischemic Digital Ulcers in Patients with Systemic Sclerosis: DUAL-1 and DUAL-2 Randomized Clinical Trials. JAMA 2016, 315, 1975–1988. [Google Scholar] [CrossRef]
- Jin, Q.; Chen, D.; Zhang, X.; Zhang, F.; Zhong, D.; Lin, D.; Guan, L.; Pan, W.; Zhou, D.; Ge, J. Medical Management of Pulmonary Arterial Hypertension: Current Approaches and Investigational Drugs. Pharmaceutics 2023, 15, 1579. [Google Scholar] [CrossRef]
- Galie, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef]
- Kumar, U.; Sankalp, G.; Sreenivas, V.; Kaur, S.; Misra, D. Prospective, open-label, uncontrolled pilot study to study safety and efficacy of sildenafil in systemic sclerosis-related pulmonary artery hypertension and cutaneous vascular complications. Rheumatol. Int. 2013, 33, 1047–1052. [Google Scholar] [CrossRef]
- Hachulla, E.; Hatron, P.Y.; Carpentier, P.; Agard, C.; Chatelus, E.; Jego, P.; Mouthon, L.; Queyrel, V.; Fauchais, A.L.; Michon-Pasturel, U.; et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: The placebo-controlled SEDUCE study. Ann. Rheum. Dis. 2016, 75, 1009–1015. [Google Scholar] [CrossRef]
- Jing, Z.C.; Yu, Z.X.; Shen, J.Y.; Wu, B.X.; Xu, K.F.; Zhu, X.Y.; Pan, L.; Zhang, Z.L.; Liu, X.Q.; Zhang, Y.S.; et al. Vardenafil in pulmonary arterial hypertension: A randomized, double-blind, placebo-controlled study. Am. J. Respir. Crit. Care Med. 2011, 183, 1723–1729. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef]
- Khanna, D.; Allanore, Y.; Denton, C.P.; Kuwana, M.; Matucci-Cerinic, M.; Pope, J.E.; Atsumi, T.; Becvar, R.; Czirjak, L.; Hachulla, E.; et al. Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): Randomised, double-blind, placebo-controlled multicentre trial. Ann. Rheum. Dis. 2020, 79, 618–625. [Google Scholar] [CrossRef]
- Distler, O.; Allanore, Y.; Denton, C.P.; Kuwana, M.; Matucci-Cerinic, M.; Pope, J.E.; Atsumi, T.; Becvar, R.; Czirjak, L.; Hachulla, E.; et al. Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): Open-label, long-term extension of a phase 2b, randomised, placebo-controlled trial. Lancet Rheumatol. 2023, 5, e660–e669. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Shillington, A.; Rich, S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation 2002, 106, 1477–1482. [Google Scholar] [CrossRef]
- Sitbon, O.; Vonk Noordegraaf, A. Epoprostenol and pulmonary arterial hypertension: 20 years of clinical experience. Eur. Respir. Rev. 2017, 26, 160055. [Google Scholar] [CrossRef] [PubMed]
- Colaci, M.; Lumetti, F.; Giuggioli, D.; Guiducci, S.; Bellando-Randone, S.; Fiori, G.; Matucci-Cerinic, M.; Ferri, C. Long-term treatment of scleroderma-related digital ulcers with iloprost: A cohort study. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 106), 179–183. [Google Scholar] [PubMed]
- Benza, R.L.; Tapson, V.F.; Gomberg-Maitland, M.; Poms, A.; Barst, R.J.; McLaughlin, V.V. One-year experience with intravenous treprostinil for pulmonary arterial hypertension. J. Heart Lung Transplant. 2013, 32, 889–896. [Google Scholar] [CrossRef]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Nathan, S.D.; Waxman, A.; Rajagopal, S.; Case, A.; Johri, S.; DuBrock, H.; De La Zerda, D.J.; Sahay, S.; King, C.; Melendres-Groves, L.; et al. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: A post-hoc analysis of the INCREASE study. Lancet Respir. Med. 2021, 9, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, T.; Tsubouchi, K.; Zhou, Q.; Chong, M.; Otsubo, K.; Isshiki, T.; Schupp, J.C.; Sato, S.; Scallan, C.; Upagupta, C.; et al. Vascular-Parenchymal Cross-Talk Promotes Lung Fibrosis through BMPR2 Signaling. Am. J. Respir. Crit. Care Med. 2023, 207, 1498–1514. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galie, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef]
- Gaine, S.; Chin, K.; Coghlan, G.; Channick, R.; Di Scala, L.; Galie, N.; Ghofrani, H.A.; Lang, I.M.; McLaughlin, V.; Preiss, R.; et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602493. [Google Scholar] [CrossRef]
- Butikofer, L.; Varisco, P.A.; Distler, O.; Kowal-Bielecka, O.; Allanore, Y.; Riemekasten, G.; Villiger, P.M.; Adler, S.; EUSTAR Collaborators. ACE inhibitors in SSc patients display a risk factor for scleroderma renal crisis—A EUSTAR analysis. Arthritis Res. Ther. 2020, 22, 59. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Badesch, D.; Chung, L.; Domsic, R.T.; Medsger, T.; Pinckney, A.; Keyes-Elstein, L.; D’Aveta, C.; Spychala, M.; White, R.J.; et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis-associated Pulmonary Arterial Hypertension: A Multicenter, Double-Blind, Randomized, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 209–221. [Google Scholar] [CrossRef]
- Medrek, S.; Melendres-Groves, L. Evolving nonvasodilator treatment options for pulmonary arterial hypertension. Curr. Opin. Pulm. Med. 2022, 28, 361–368. [Google Scholar] [CrossRef]
- Johnson, S.R.; Granton, J.T.; Tomlinson, G.A.; Grosbein, H.A.; Le, T.; Lee, P.; Seary, M.E.; Hawker, G.A.; Feldman, B.M. Warfarin in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. A Bayesian approach to evaluating treatment for uncommon disease. J. Rheumatol. 2012, 39, 276–285. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grunig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef]


| PH Animal Model | Vessel Occlusion | Elevation of PA Pressure | RV Remodeling | Interstitial Lung Disease | Perivascular Inflammation | Spontaneous Regression | Systemic Features | Plexiform Lesions | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Monocrotaline ® | +++ | ++++ | +++ | − | +++ | + | − | +++ | [59] |
| Sugen/Hypoxia ® | ++++ | ++++ | +++ | − | +++ | − | emphysema | +++ | [60] |
| Sugen/Hypoxia (M) | ++ | ++ | ++ | − | ++ | + | − | − | [30] |
| BMPR2+/− (M) | ++ | + | ++ | − | ++ | − | − | + | [61] |
| Fli-1/Klf5 het. (M) | + | n.r. | n.r. | fibrotic | ++ | − | skin fibrosis | − | [62] |
| Fra-2 (M) | ++ | + | ++ | fibrotic | ++ | − | skin fibrosis | − | [63] |
| TNF-Tg (M) | ++++ | +++ | +++ | inflammatory | ++++ | − | arthritis | + | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahi, M.; Li, C.; Wang, G.; Korman, B.D. Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: From Bedside to Bench and Back Again. Int. J. Mol. Sci. 2024, 25, 4728. https://doi.org/10.3390/ijms25094728
Bahi M, Li C, Wang G, Korman BD. Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: From Bedside to Bench and Back Again. International Journal of Molecular Sciences. 2024; 25(9):4728. https://doi.org/10.3390/ijms25094728
Chicago/Turabian StyleBahi, Milan, Christine Li, Gaochan Wang, and Benjamin D. Korman. 2024. "Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: From Bedside to Bench and Back Again" International Journal of Molecular Sciences 25, no. 9: 4728. https://doi.org/10.3390/ijms25094728
APA StyleBahi, M., Li, C., Wang, G., & Korman, B. D. (2024). Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: From Bedside to Bench and Back Again. International Journal of Molecular Sciences, 25(9), 4728. https://doi.org/10.3390/ijms25094728