Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs

Abstract
:Introduction
Materials and Methods
Mathematical Definition of the Calculated Molecular Descriptors
Molecular vector space
Total quadratic indices; [qk(x)]
= Lii if i = j
= 0 otherwise


{kind=link}
{kind=link}
{kind=link}
| Benzene | ||||||||
| q0(x) | q1(x) | q2(x) | q3(x) | q4(x) | q5(x) | q6(x) | q7(x) | |
| P | 41.5014 | 124.5042 | 373.5126 | 1120.5378 | 3361.6134 | 10084.8402 | 30254.5206 | 90763.5618 |
| MA | 41.5014 | 124.5042 | 373.5126 | 1120.5378 | 3361.6134 | 10084.8402 | 30254.5206 | 90763.5618 |
| MB | 41.5014 | 124.5042 | 373.5126 | 1120.5378 | 3361.6134 | 10084.8402 | 30254.5206 | 90763.5618 |
| Acetylsalicylic acid | ||||||||
| Total | ||||||||
| q0(x) | q1(x) | q2(x) | q3(x) | q4(x) | q5(x) | q6(x) | q7(x) | |
| P | 102.4477 | 268.8912 | 873.5982 | 2566.8034 | 8381.4114 | 25593.6122 | 83330.7872 | 260026.931 |
| MA | 102.4477 | 268.8912 | 873.5982 | 2549.8376 | 8284.7898 | 25063.374 | 81351.7828 | 250745.988 |
| MB | 102.4477 | 268.8912 | 873.5982 | 2566.5118 | 8389.425 | 25513.2092 | 83389.772 | 258104.308 |
| Local | ||||||||
| Eq0(x) | Eq1(x) | Eq2(x) | Eq3(x) | Eq4(x) | Eq5(x) | Eq6(x) | Eq7(x) | |
| P | 40.1956 | 58.3597 | 265.963 | 510.2749 | 2171.4817 | 4947.1654 | 19328.9482 | 49869.8377 |
| MA | 40.1956 | 58.3597 | 265.963 | 500.226 | 2133.2198 | 4618.7534 | 18773.2472 | 44486.7656 |
| MB | 40.1956 | 58.3597 | 265.963 | 508.5631 | 2201.8503 | 4802.1696 | 19870.6695 | 47162.9747 |
| Hq0(x) | Hq1(x) | Hq2(x) | Hq3(x) | Hq4(x) | Hq5(x) | Hq6(x) | Hq7(x) | |
| P | 4.84 | 6.974 | 10.626 | 33.682 | 67.54 | 270.578 | 670.604 | 2600.972 |
| MA | 4.84 | 6.974 | 10.626 | 33.682 | 67.54 | 269.632 | 647.306 | 2589.686 |
| MB | 4.84 | 6.974 | 10.626 | 33.682 | 67.54 | 271.766 | 653.092 | 2639.868 |
Local approach (local invariant) of the quadratic indices; [qkL(x)]
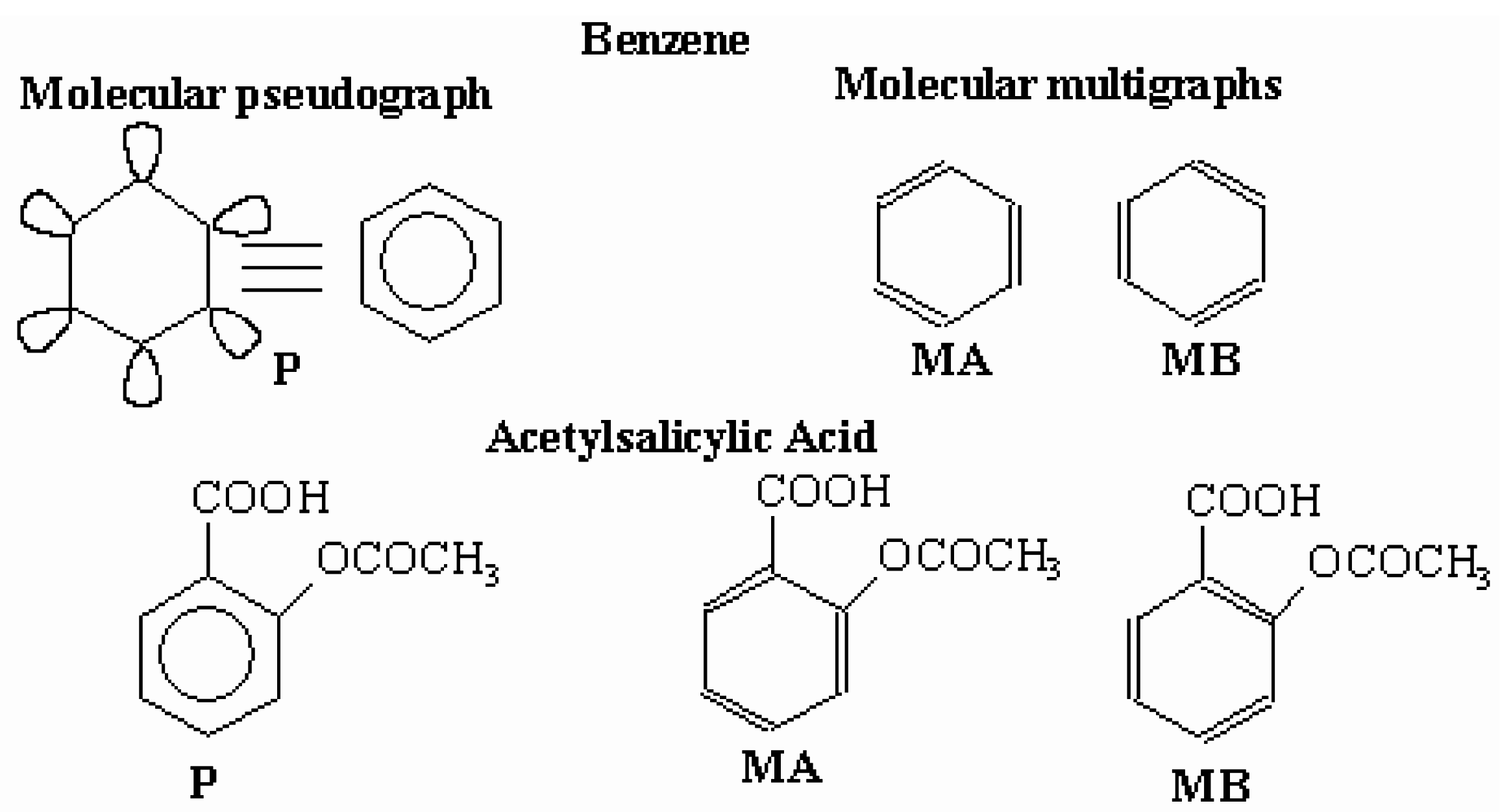
 Acetylsalicylic Acid Molecular structure |  Molecular Pseudograph (G)(Suppressed Hydrogen Atom) | X=[O1 O2 C3 C4 C5 C6 C7 C8 C9 O10 C11 O12 C13]
Molecular Vector: X∈ ℜ13 and ℜ13∈E; E: Molecular Vectorial Space In the definition of the X, as molecular vector, the chemical symbol of the element is used to indicate the corresponding electronegativity value. That is: if we write O it means χ(O), oxygen Mulliken electronegativity or some atomic property, which characterizes each atom in the molecule. So, if we use the canonic bases of R13, the coordinates of any vector X coincide with the components of that molecular vector Xt =[3.17 3.17 2.63 2.63 2.63 2.63 2.63 2.63 2.63 3.17 2.63 3.17 2.63] Xt = transposed of X and it means the vector of the coordinates of X in the Canonical base of R13 (a row Matrix) X: vector of coordinates of X in the Canonical base of R13 (a columns matrix) |
| = XtM0X=102.4472 | ||
| = XtM1X=268.8912 | ||
| = XtM2X=373.5982 | M1 (G) : Molecular Pseudograph’s Atom Adjacency Matrix | |
| = XtM3X=2566.8034 | ||
| = XtM4X=8381.1414 | ||
| = XtM5X=25593.612 | ||
=1/2kaij if either vi or vj is contained in the specific fragment but not both at the same time
=0 otherwise
The TOMO-COMD Software
- Draw the molecular pseudographs for each molecule of the data set, using the software drawing mode. This procedure is carried out by a selection of the active atom symbol belonging to different groups of the periodic table. The multiples edges and loops are edited with a right mouse click,
- Use appropriated atom weights in order to differentiate the molecular atoms. In this work, we used as atomic property the electronegativity of Mulliken [48] for each kind atom,
- Compute the total and local quadratic indices of the molecular pseudograph’s atom adjacency matrix. They can be carried out in the software calculation mode, which you can select the atomic properties and the family descriptor previously to calculate the molecular indices. This software generate a table in which the rows correspond to the compounds and columns correspond to the total and local quadratic indices or any others family molecular descriptors implemented in this program,
- Find a QSPR/QSAR equation by using statistical techniques, such as multilinear regression analysis (MRA), Neural networks, linear discrimination analysis, and so on. That is to say, we can find a quantitative relation between a property P and the quadratic indices having, for instance, the following appearance:where P is the measurement of the property, qk(x) [or qkL(x)] is the kth total [or local] quadratic indices, an the ak’s are the coefficients obtained by the linear regression analysis.P=a0q0(x) + a1q1(x) + a2q2(x) +….+ akqk(x) + c
- Test the robustness and predictive power of the QSPR/QSAR equation by using internal and external cross-validation techniques,
- Develop a structural interpretation of obtained QSAR/QSPR model using quadratic indices as molecular descriptors.
- (1)
- qk(x) and qkH(x) are the k-th total quadratic indices calculated using the k-th power of the matrices [Mk(G)] of the molecular pseudograph (G) considering and not considering hydrogen atoms, respectively.
- (2)
- EqkL(x) [or EqkLH(x)] and H qkk(x) are the k-th local quadratic indices calculated using a k-th power of the local matrices [MkL(G, Fi)] of the molecular pseudograph (G) not considering (or considering) hydrogen atoms for heteroatoms (S,N,O) and hydrogen bonding heteroatoms (S,N,O), respectively.
Caco-2 Cell Permeation Coefficients
Statistical Analysis
Results and Discussion
Quantitative Structure Permeability Relationships
+0.004175 (± 1.618x10-3).q0H(x)
Log Pcaco-2 = -3.16658 (± 0.194)-0.00291(± 0.238x10-4)..Hq5L(x)
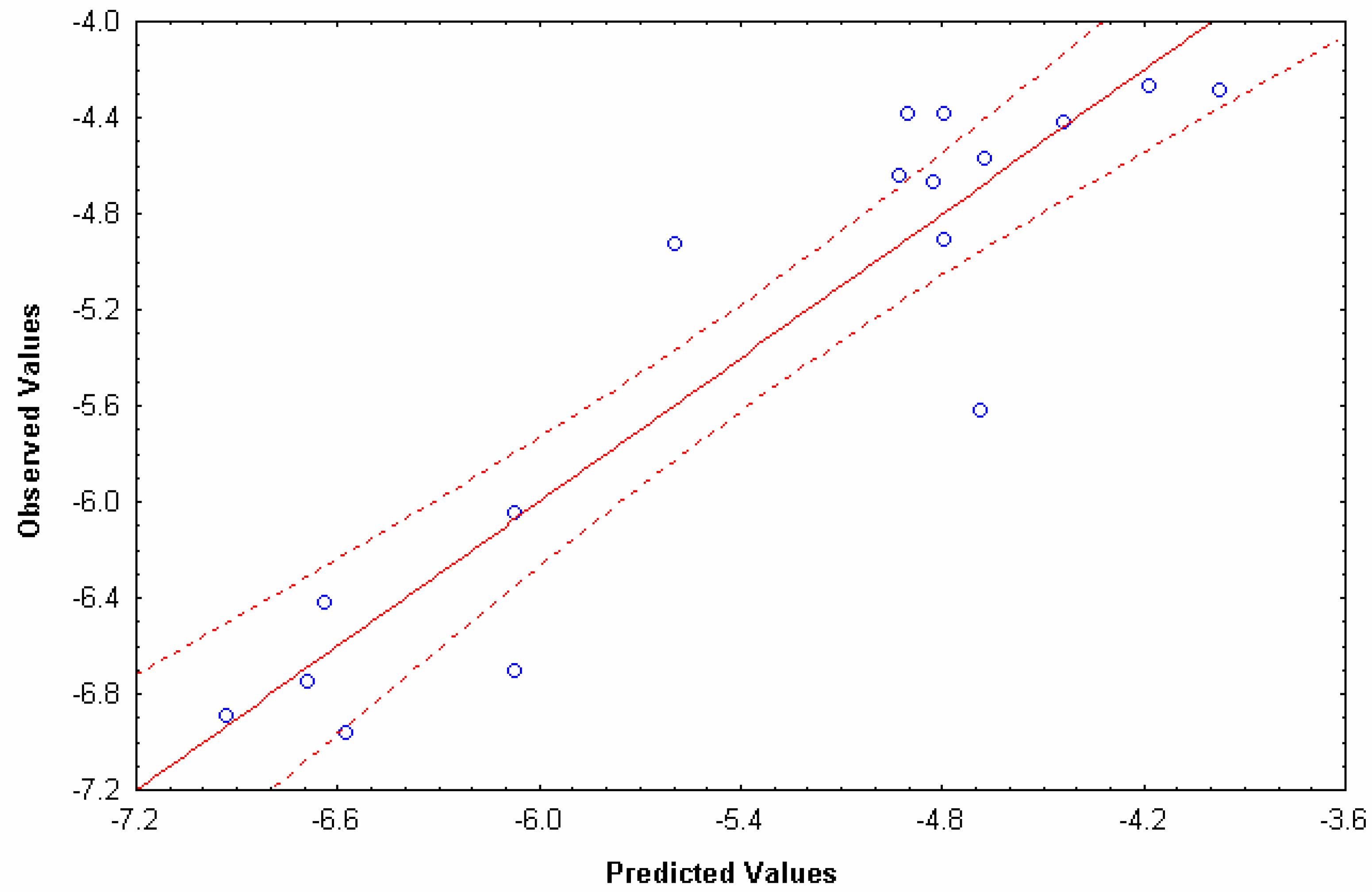
N=16 R=0.96 RCV=0.93 s=0.32 RMSECV=0.35 F(1,14)=149.45 p<0.0000
| Compounds* | Obs.a | Cal.b | Res.c | CV-resd | Cal.e | Res.c | CV-resd | q0H(x)f | Hq5L(x)g |
|---|---|---|---|---|---|---|---|---|---|
| Training set | |||||||||
| Alprenolol | -4.378 | -4.795 | 0.417 | 0.458 | -4.550 | 0.172 | 0.191 | 235.7602 | 475.2 |
| Testosterone | -4.286 | -3.973 | -0.313 | -0.384 | -3.829 | -0.457 | -0.574 | 287.0389 | 227.458 |
| Metoprolol | -4.569 | -4.675 | 0.106 | 0.116 | -4.550 | -0.019 | -0.021 | 264.4829 | 475.2 |
| Salicylic acid | -4.924 | -5.598 | 0.674 | 1.028 | -4.868 | -0.056 | -0.061 | 107.605 | 584.386 |
| Propranol | -4.378 | -4.905 | 0.527 | 0.572 | -4.691 | 0.313 | 0.343 | 237.8371 | 523.732 |
| Corticosterone | -4.263 | -4.185 | -0.078 | -0.096 | -4.297 | 0.034 | 0.039 | 330.6505 | 388.212 |
| Warfarin | -4.417 | -4.437 | 0.020 | 0.023 | -4.191 | -0.226 | -0.264 | 249.0567 | 351.758 |
| Hydrocortisone | -4.668 | -4.830 | 0.162 | 0.199 | -5.112 | 0.444 | 0.475 | 340.6994 | 668.118 |
| Dexamethasone | -4.903 | -4.793 | -0.110 | -0.144 | -5.154 | 0.251 | 0.268 | 358.0644 | 682.638 |
| Acetylsalicilic acid* | -5.62 | -4.688 | -0.932 | -1.409 | - | - | - | 141.1677 | 270.578 |
| Atenolol | -6.7 | -6.076 | -0.624 | -0.693 | -6.089 | -0.611 | -0.678 | 239.4811 | 1003.904 |
| Terbutaline | -6.42 | -6.641 | 0.221 | 0.268 | -6.535 | 0.115 | 0.136 | 193.9415 | 1156.98 |
| Mannitol | -6.744 | -6.693 | -0.051 | -0.064 | -6.476 | -0.269 | -0.315 | 169.5548 | 1136.696 |
| Sulphasalasine | -6.886 | -6.936 | 0.050 | 0.073 | -7.262 | 0.376 | 0.536 | 270.0324 | 1406.702 |
| Practolol | -6.046 | -6.073 | 0.027 | 0.030 | -6.086 | 0.040 | 0.045 | 239.4811 | 1002.892 |
| Olsalazine | -6.959 | -6.577 | -0.382 | -0.457 | -6.569 | -0.390 | -0.464 | 216.3878 | 1168.772 |
| Felodipine | -4.644 | -4.929 | 0.285 | 0.310 | -4.929 | 0.285 | 0.307 | 280.0887 | 605.462 |
| External test set | |||||||||
| Compounds | Obs.h | Cal.b | Res.c | Cal.e | Res.c | q0H(x)f | Hq5L(x)g | ||
| Cumarin | -4.11 | -4.149 | 0.039 | -3.167 | -0.943 | 111.3899 | 0 | ||
| Theophyline | -4.35 | -5.328 | 0.978 | -4.653 | 0.303 | 128.9517 | 510.576 | ||
| Epinephrine | -6.02 | -6.699 | 0.679 | -6.438 | 0.418 | 160.7477 | 1123.914 | ||
| Guanoxan* | -4.71 | -6.876 | 2.166 | -6.687 | 1.977 | 168.4735 | 1209.406 | ||
| Guanabenz* | -4.14 | -7.011 | 2.871 | -6.675 | 2.535 | 133.7708 | 1205.226 | ||
| Lidocaine | -4.21 | -5.081 | 0.871 | -4.832 | 0.622 | 224.2233 | 572.088 | ||
| Tiacrilast | -4.90 | -4.482 | -0.418 | -3.894 | -1.006 | 178.2154 | 249.766 | ||
| Imipramine | -4.26 | -3.535 | -0.725 | -3.167 | -1.093 | 258.4389 | 0 | ||
| Furosemide | -6.09 | -8.424 | 2.334 | -8.741 | 2.651 | 212.1532 | 1914.814 | ||
| Sulpiride | -6.16 | -7.328 | 1.168 | -7.763 | 1.603 | 277.3639 | 1578.896 | ||
| Nitrendipine | -4.77 | -4.850 | 0.080 | -4.873 | 0.103 | 287.6154 | 586.102 | ||
| Fleroxacin | -4.81 | -4.055 | -0.755 | -3.951 | -0.859 | 292.165 | 269.5 | ||
| Diltiazem | -4.31 | -3.236 | -1.074 | -3.167 | -1.143 | 330.0333 | 0 | ||
| Verapamil | -4.58 | -2.853 | -1.727 | -3.167 | -1.413 | 421.7297 | 0 | ||
| Mibefradil | -4.87 | -4.150 | -0.720 | -4.828 | -0.042 | 446.2316 | 570.702 | ||
| Bosentan | -5.98 | -5.270 | -0.710 | -6.029 | 0.049 | 420.3623 | 983.422 | ||
| Proscillaridin* | -6.20 | -4.662 | -1.538 | -5.634 | -0.566 | 486.3382 | 847.66 | ||
| Ceftriaxone | -6.88 | -7.368 | 0.488 | -8.030 | 1.150 | 321.3851 | 1670.482 | ||
| Remikiren | -6.13 | -6.651 | 0.521 | -8.327 | 2.197 | 553.2348 | 1772.76 | ||
| Squinavir | -6.26 | -8.734 | 2.474 | -9.320 | 3.060 | 254.6519 | 2113.892 | ||


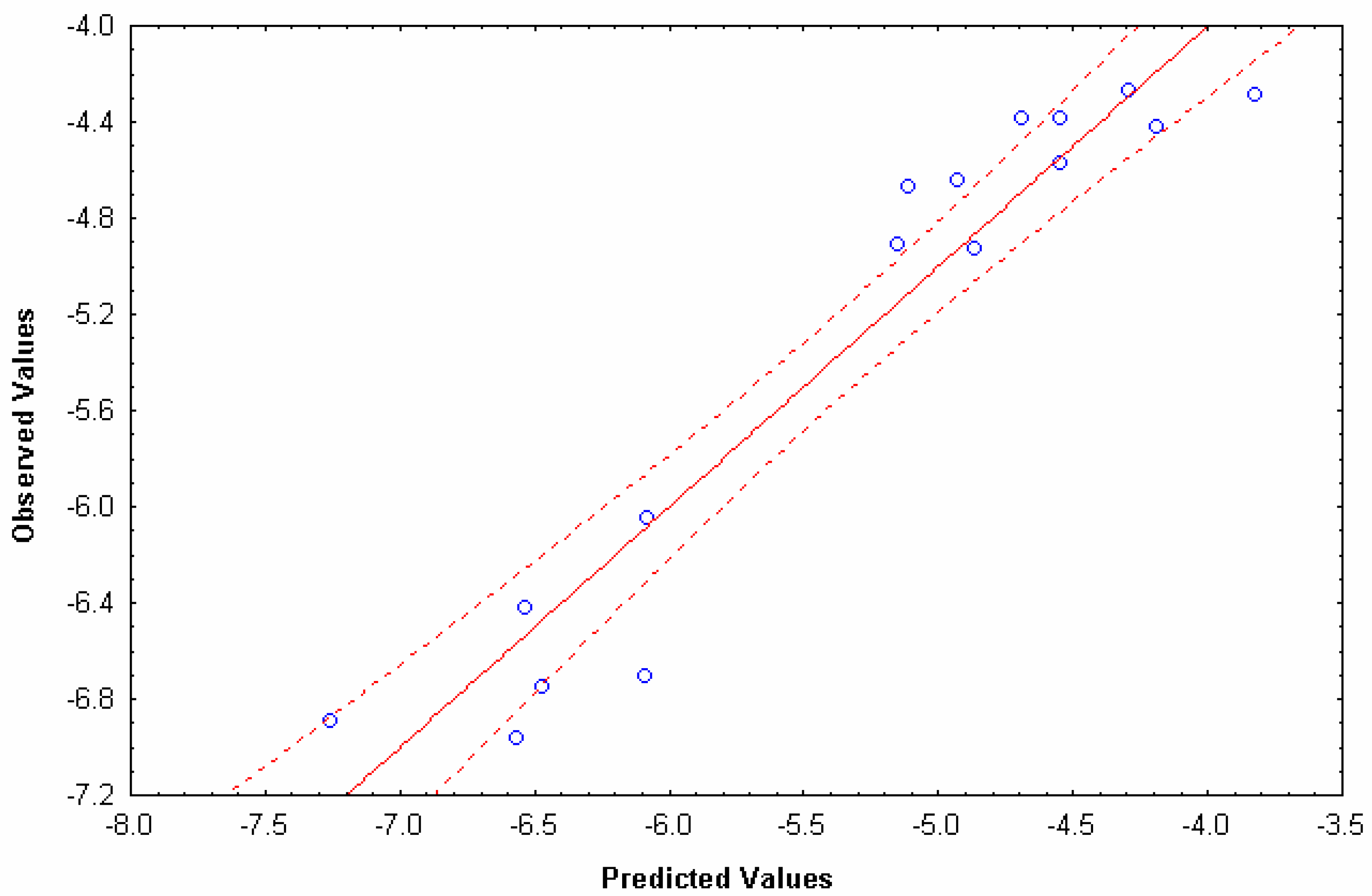
N=5 R=0.91 F(1, 3)= 14.079 s=0.55 p<0.0000
N=6 R=0.95 F(1, 4)= 37.784 s=0.32 p<0.0000
N=6 R=0.99 F(1, 4)= 300.81 s=0.14 p<0.0000
| Compounds | Obs. | Cal. | Res. | CV-res | Eq0L(x) | Hq3L(x) | Hq4L(x) |
|---|---|---|---|---|---|---|---|
| Anionic compounds (-) | |||||||
| Salicylic acid | -4.924 | -4.693 | -0.231 | -0.431 | 30.1467 | 64.988 | 158.466 |
| Warfarin | -4.417 | -5.187 | 0.770 | 1.064 | 40.1956 | 31.306 | 90.684 |
| Acetylsalicylic acid | -5.62 | -5.187 | -0.433 | -0.599 | 40.1956 | 33.682 | 67.54 |
| Sulphasalazine | -6.886 | -7.032 | 0.146 | 0.343 | 77.7682 | 130.746 | 332.046 |
| Olsalazine | -6.959 | -6.707 | -0.252 | -0.425 | 71.1512 | 129.976 | 316.932 |
| Neutral compounds (0) | |||||||
| Testosterone | -4.286 | -3.881 | -0.405 | -0.727 | 20.0978 | 30.36 | 71.434 |
| Corticosterone | -4.263 | -4.325 | 0.062 | 0.084 | 40.1956 | 59.774 | 128.832 |
| Hydrocortisone | -4.668 | -5.030 | 0.362 | 0.436 | 50.2445 | 91.08 | 220 |
| Dexamethasone | -4.903 | -5.066 | 0.163 | 0.197 | 65.5326 | 91.08 | 224.708 |
| Mannitol | -6.7445 | -6.466 | -0.278 | -1.281 | 60.2934 | 180.268 | 405.768 |
| Felodipine | -4.644 | -4.739 | 0.095 | 0.116 | 63.6245 | 50.094 | 182.424 |
| Cationic compounds (+) | |||||||
| Alprenol | -4.378 | -4.394 | 0.016 | 0.024 | 25.5267 | 72.776 | 190.036 |
| Metoprol | -4.569 | -4.394 | -0.175 | -0.267 | 35.5756 | 72.776 | 190.036 |
| Propranol | -4.378 | -4.546 | 0.168 | 0.239 | 25.5267 | 77.616 | 200.662 |
| Atenolol | -6.7 | -6.601 | -0.099 | -0.167 | 41.0045 | 143 | 371.624 |
| Terbutaline | -6.42 | -6.514 | 0.094 | 0.149 | 35.5756 | 140.228 | 381.326 |
| Practolol | -6.046 | -6.043 | -0.003 | -0.004 | 41.0045 | 125.246 | 349.602 |
Interpretation of QSPerR Models
| Tolerance | Corr. Partial | |
| Heq5L(x) | 0.981901 | -0.90851 |
| eq0H(x) | 0.981901 | 0.56783 |
| Correlation Matrix | ||
| Heq5L(x) | eq0H(x) | |
| Heq5L(x) | 1 | 0.134534 |
| eq0H(x) | 0.134534 | 1 |
Virtual Screening and relationship of human intestinal absorption and Caco-2 cell permeability
| Compounds | Cal. | Obs.c | % Absorbed | Ref. | Hq5L(x) | q0H(x) |
|---|---|---|---|---|---|---|
| Acebutolol | 0.53a | 0.51 | 90 | 6 | 1067.836 | 311.0776 |
| Acetylsalicylic acid | 20.50b | 30.67 | 68 | 62 | 270.578 | 141.1677 |
| 9.09 | 100 | 6 | ||||
| 2.40 | 100 | 7 | ||||
| Alprenolol | 28.19a | 40.50 | 93 | 7 | 475.2 | 235.7602 |
| 25.30 | 93 | 6 | ||||
| Aminopyrine | 163.99b | 36.50 | 100 | 6 | 0 | 198.5353 |
| Atenolol | 0.84b | 4.00 | 50 | 60 | 1003.904 | 239.4811 |
| 1.16 | 40-70, 50 | 63 | ||||
| 0.53 | 50 | 6 | ||||
| 0.20 | 50 | 7 | ||||
| 0.13 | 40 | 64 | ||||
| Penicilin G | 5.40b | 1.96 | 30 | 62 | 700.128 | 254.6519 |
| Caffeine | 98.53b | 84.29 | 100 | 63 | 0 | 145.5486 |
| 50.50 | 100 | 62 | ||||
| 30.80 | 100 | 6 | ||||
| 21.40 | 100 | 60 | ||||
| Chloramphenicol | 2.49a | 20.60 | 90 | 62 | 837.386 | 213.2682 |
| Cimetidine | 0.12b | 3.06 | 62 | 62 | 1251.47 | 184.9905 |
| 1.37 | 95 | 6 | ||||
| Clonidine | 3.13a | 30.10 | 95 | 62 | 803.264 | 140.0988 |
| 21.80 | 100 | 6 | ||||
| Corticosterone | 50.50a | 21.20 | 100 | 6 | 388.212 | 330.6505 |
| Desipramine | 48.65b | 24.40 | 95 | 6 | 288.97 | 241.842 |
| 21.60 | 100 | 62 | ||||
| Dexamethasone | 16.11b | 23.40 | 92 | 62 | 682.638 | 358.0644 |
| 12.50 | 100 | 7 | ||||
| 12.20 | 100 | 6 | ||||
| Diazepam | 172.00b | 70.97 | 100 | 62 | 0 | 203.4971 |
| 33.40 | 100 | 6 | ||||
| Felodipine | 11.77b | 22.70 | 100 | 7 | 605.462 | 280.0887 |
| Fluconazole | 46.07b | 29.80 | 100 | 62 | 263.45 | 221.1982 |
| Ganciclovir | 0.07b | 2.67 | 8 | 61 | 1361.932 | 192.5122 |
| 0.38 | 3 | 6 | ||||
| Hydrocortisone | 14.80b | 44.67 | 95 | 65 | 668.118 | 340.6994 |
| 35.40 | 80 | 61 | ||||
| 21.50 | 89 | 7 | ||||
| 14.00 | 89 | 6 | ||||
| 12.19 | 80, 89, 95 | 63 | ||||
| Ibuprofen | 39.41b | 52.50 | 100 | 62 | 250.14 | 197.1375 |
| Imipramine | 291.70b | 14.10 | 100 | 62 | 0 | 258.4389 |
| Indomethacin | 80.96b | 20.40 | 100 | 6 | 235.62 | 263.4856 |
| Labetalol | 0.08b | 9.31 | 90 | 6 | 1494.878 | 288.5856 |
| Mannitol | 0.33a | 3.23 | 17 | 61 | 1136.696 | 169.5548 |
| 1.17 | 5, 16, 17 | 63 | ||||
| 0.83 | 5 | 65 | ||||
| 0.65 | 16 | 62 | ||||
| 0.50 | 16 | 60 | ||||
| 0.38 | 16 | 6 | ||||
| 0.18 | 16 | 7 | ||||
| Meloxicam | 2.34a | 19.50 | 90 | 6 | 846.714 | 227.8551 |
| Metoprolol | 21.13b | 27.00 | 95 | 7 | 475.2 | 264.4829 |
| 23.70 | 95 | 6 | ||||
| 18.00 | 95 | 63 | ||||
| Nadolol | 2.37b | 4.50 | 35 | 60 | 904.75 | 289.0518 |
| Noloxone | 13.21b | 28.20 | 91 | 62 | 582.604 | 278.6856 |
| Naproxen | 38.51b | 74.17 | 100 | 61 | 250.14 | 194.7433 |
| Nevirapine | 12.27a | 30.10 | 90 | 6 | 599.236 | 203.278 |
| Nicotine | 100.67b | 19.40 | 100 | 6 | 0 | 147.7868 |
| Phenytoin | 0.61a | 89.83 | 100 | 65 | 1046.672 | 192.7891 |
| 26.70 | 90 | 6 | ||||
| Pindolol | 0.46b | 16.70 | 95 | 6 | 1085.788 | 224.5922 |
| Piroxicam | 1.44b | 35.60 | 100 | 6 | 890.208 | 228.9639 |
| Practolol | 0.84b | 0.90 | 100 | 7 | 1002.892 | 239.4811 |
| Progesterone | 481.44b | 78.93 | 100 | 61 | 0 | 310.5527 |
| Propranolol | 20.36a | 41.90 | 90 | 7 | 523.732 | 237.8371 |
| 34.43 | 90 | 63 | ||||
| 27.50 | 90 | 62 | ||||
| 21.80 | 90 | 6 | ||||
| 14.80 | 90 | 60 | ||||
| Salicylic acid | 13.56a | 41.90 | 100 | 60 | 584.386 | 107.605 |
| 22.00 | 100 | 6 | ||||
| 11.90 | 100 | 7 | ||||
| Sucrose | 0.07b | 0.71 | 42 | 63 | 1557.072 | 300.0207 |
| Sumatriptan | 0.24b | 3.00 | 55 | 62 | 1225.466 | 240.6692 |
| Telmisartan | 112.00a | 15.10 | 90 | 6 | 269.39 | 415.2711 |
| Tenidap | 17.85a | 51.20 | 90 | 62 | 543.4 | 196.2092 |
| Terbutaline | 0.29a | 1.04 | 25-80, 73 | 63 | 1156.98 | 193.9415 |
| 0.47 | 73 | 6 | ||||
| 0.38 | 73 | 7 | ||||
| Testosterone | 106.32b | 72.27 | 100 | 62 | 227.458 | 287.0389 |
| 51.80 | 100 | 7 | ||||
| 44.50 | 100 | 63 | ||||
| 24.90 | 100 | 6 | ||||
| Timolol | 14.01b | 12.80 | 72 | 6 | 546.766 | 263.7501 |
| Valproic acid | 25.89b | 48.00 | 100 | 62 | 249.194 | 152.873 |
| Warfarin | 36.58b | 38.30 | 98 | 7 | 351.758 | 249.0567 |
| 21.10 | 98 | 6 | ||||
| Ziprasidone | 15.53a | 12.30 | 60 | 62 | 564.168 | 293.4675 |
| Cephalexin | 0.23b | 2.69 | 100 | 63 | 1261.81 | 255.2408 |
| 0.18 | 95 | 64 | ||||
| 0.50 | 100 | 60 | ||||
| L-Phenylalanine | 6.23a | 29.50 | 100 | 65 | 700.348 | 141.0188 |
| 6.91 | 100 | 63 | ||||
| Antipyrine | 107.98b | 49.01 | 97 | 63 | 0 | 155.0726 |
| Guanabenz | 0.21a | 20.90 | 79 | 60 | 1205.226 | 133.7708 |
| Glycine | 30.93a | 80.00 | 100 | 62 | 461.362 | 63.5605 |
| D-Phe-L-Pro | 5.62a | 44.30 | 100 | 62 | 715.704 | 224.9611 |
| Gabapentin | 5.17b | 4.33 | 74 | 65 | 563.728 | 170.0588 |
| 1.50 | 36 | 65 | ||||
| BVaraU | 0.43a | 4.00 | 82 | 60 | 1099.494 | 217.7747 |
| Pravastatin | 2.19a | 2.30 | 34 | 60 | 856.548 | 398.831 |
| Amoxicillin | 0.06b | 0.80 | 100 | 60 | 1534.962 | 274.9697 |
| 0.33 | 100 | 63 | ||||
| SQ-29852 | 2.84a | 0.02 | 60 | 60 | 817.476 | 374.5622 |
| Trovafloxacin | 5.40b | 30.23 | 88 | 62 | 783.772 | 303.8246 |
| Scopolamine | 94.77b | 11.80 | 100 | 6 | 181.786 | 248.2549 |
| Ziduvudine | 0.00b | 6.93 | 100 | 6 | 1937.496 | 204.2691 |
| Taurocholic acid | 0.43b | 4.02 | 100 | 63 | 1499.63 | 459.8587 |
| Acyclovir | 0.25a | 2.00 | 30 | 62 | 1179.332 | 165.8664 |
| 0.25 | 20 | 6 | ||||
| Methotrexate | 0.00b | 1.20 | 20 | 62 | 2153.426 | 338.4937 |
| Glutamine | 0.19a | 0.85 | 60-90 | 63 | 1221.484 | 123.989 |
| Enaprilate | 9.42a | 0.62 | 10 | 63 | 638.748 | 330.1203 |
| Hidrochlorothiazide | 0.00b | 0.51 | 90 | 6 | 2336.84 | 164.2368 |
| Ranitidine | 2.65b | 0.49 | 50 | 6 | 824.978 | 254.0701 |
| Sulphasalazine | 0.12b | 0.30 | 13 | 6 | 1406.702 | 270.0324 |
| 0.13 | 13 | 7 | ||||
| Doxorubicin | 0.08b | 0.16 | 5 | 62 | 1761.694 | 433.4031 |
| Olsalazine | 0.27b | 0.11 | 2 | 7 | 1168.772 | 216.3878 |
| Lisinopril | 0.14a | 0.05 | 25 | 60 | 1271.886 | 356.9861 |
Conclusions
References
- Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Anonymous. Waiver of in vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. 2000. Available from http://www.fda.gov/cder/OPS/BCS_guidance.htm. [Google Scholar]
- Rodríguez, A. D. Rational High Throughput Screening in Preclinical Drug Metabolism. Med Chem Res. 1998, 8, 422–433. [Google Scholar]
- Artusson, P. Cell Cultures as Models for Drug Absorption Across the Intestinal Mucosa. Cri. Rev. Ther. Carrier Syst. 1991, 8, 305–330. [Google Scholar]
- Quaroni, A.; Hochman, J. Development of Intestinal Cell Culture Models for Drug Transport and Metabolism Studies. Adv. Drug Del. Rev. 1996, 22, 3–52. [Google Scholar] [CrossRef]
- Yazdanian, M.; Glynn, S. L.; Wright, J. L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P.; Karlsson, J. Correlation between Oral Drug Permeability Coefficients in Human Intestinal Epithelial (Caco-2) Cells. Biochem. Biophys Res. Com. 1991, 175, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, G.; Alsenz, J.; Van de Waterbeemd, H.; Folkers, G. Estimation of Permeability by Passive Diffusion Through Caco-2 Cell Monolayer Using the Drugs´ Lipophilicity and Molecular Weight. Eur. J. Pharm. Sci. 1998, 6, 313–319. [Google Scholar] [CrossRef]
- Yazdanian, M.; Glynn, S. L.; Wright, J. L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 Monolayer in Experimental and Theoretical Predictions of Drug Transport. Adv. Drug Del. Rev. 1996, 22, 67–84. [Google Scholar] [CrossRef]
- Delie, F.; Rubas, W. A. Human Colonic Cell Line Sharing Similarities with Enterocytes as a Model to Examine Oral Absorption. Crit. Rev. Ther. Drug Carrier Syst. 1997, 14, 221–286. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I. J.; Raub, T. J.; Borchardt, R. T. Characterization of the Human Colon Carcinoma Cell Line (Caco-2) as a Model System for Intestinal Permeability. Gastroenterology. 1989, 96, 736–749. [Google Scholar] [PubMed]
- Anderle, P.; Niederer, E.; Rubas, W.; Hilgendorf, C.; Spahn-Langguth, P. P-glicoprotein (P-gp) mediated efluxx in caco-2 cell monolayers: The Influence of Culturing Condition and Drug Exposure on P-gp Expression Levels. J. Pharm. Sci. 1998, 87, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Raevsky, O. A. Raevsky, Estimation of Caco-2 Cell Permeability Using Calculated Molecular Descriptors. Quant. Struct-Act. Relat. 1996, 15, 480–490. [Google Scholar] [CrossRef]
- Ren, S.; Lien, E. J. Caco-2 Cell Permeability vs Human Gastro-Intestinal Absorption: QSPR Analysis. Prog. Drug Res. 2000, 54, 3–23. [Google Scholar]
- Van der Waterbeemd, H.; Smith, D. A.; Jones, B. C. Lipophilicity in PK Desing: Methyl, Ethyl, Futile. J. Comp-Aided Mol. Des. 2001, 15, 273–286. [Google Scholar] [CrossRef]
- Dressman, J. B.; Amidon, G. L.; Fleisher, D. Absorption Potential: Estimating the Fraction Absorbed for Orally Administered Compounds. J. Pharm. Sci. 1985, 74, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, H. W.; Steinbaugh, B. A.; Stewart, B. H.; Chan, O. H.; Schmid, H. L.; Schroeder, R.; Ryan, M. J.; Keiser, J.; Taylor, M. D.; Blankley, C. J.; Kalttenbronn, J. S.; Wright, J.; Hicks, J. Evaluation of Physicochemical Parameters Important to the Oral Bioavailability of Peptide-Like Compounds: Implications for the Synthesis of Renin Inhibitors. J. Med. Chem. 1995, 38, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Palm, K.; Luthman, K.; Ungell, A. L.; Strandlund, G.; Artursson, P. Correlation of Drug Absorption with Molecular Surface Properties. J Pharm Sci. 1996, 85, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M. H.; Chadha, H. S.; Mitchell, R. C. Hydrogen Bonding. 33. Factors that Influence the Distribution of Solutes between Blood and Brain. J. Pharm. Sci. 1994, 83, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Basak, S. C.; Gute, B. D.; Drewes, E. R. Predicting Blood-Brain Transport of Drugs: A Computational Approach. Pharm. Res. 1996, 13, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Potts, R. O.; Guy, R. H. A Predictive Algorithm for Skin Perme-ability: The Effects of Molecular Size and Hydrogen Bond Activity. Pharm. Res. 1995, 12, 1628–1663. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, F.; Topliss, J. G. Unified Model for the Corneal Permeability of Related and Diverse Compounds with Respect to Their Physico-chemical Properties. J. Pharm. Sci. 1996, 85, 819–823. [Google Scholar] [CrossRef]
- Gobburu, J. V. S.; Shelver, W. H. Quantitative Structure-Pharmaco-kinetic Relationships (QSPR) of Beta Blockers Derived Using Neural Networks. J. Pharm. Sci. 1995, 84, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Wessel, M. D.; Jurs, P. C.; Tolan, J. W.; Muskal, S. M. Prediction of Human Intestinal Absorption of Drug Compounds from Molecular Structure. J. Chem. Inf. Comput. Sci. 1998, 38, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Clark, D. E.; Pickett, D. S. Computational Methods for the Prediction of ‘Drug-Likeness’. Drug Disc. Today. 2000, 5, 49–58. [Google Scholar] [CrossRef]
- Fujiwara, S-I.; Yamashita, F.; Hashida, M. Prediction of Caco-2 Cell Permeability Using a Combination of MO-Calculation and Neural Network. Int. J. Pharm. 2002, 237, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Han, Y.; Hopfinger, J. Predicting Caco-2 Cell Permeation Coefficients of Organic Molecules Using Membrane-Interaction QSAR Analysis. J. Chem. Inf. Comput. Sci. 2002, 42, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Van der Waterbeemd, H.; Kansy, M. Hydrogen-bonding Capacity Permeability Using Calculated Molecular Descriptors. Quant. Struct.-Act Relat. 1992, 15, 480–490. [Google Scholar] [CrossRef]
- Krarup, H.; Christensen, T. I.; Hovgaard, L.; Frokjaer, S. Predicting Drug Absortion From Molecular Surface Properties Based on Molecular Dynamics Simulations. Pharm. Res. 1998, 15, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Osterber, T.; Artursson, P. Theoretical Calculation and Prediction of Caco-2 Cell Permeability Using MolSurf Parameterization and PLS Statistics. Pharm. Res. 1997, 14, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Diudea, M. V. (Ed.) QSPR/QSAR Studies by Molecular Descriptors; Nova Science: Huntington, N.Y., 2001.
- Ivanciuc, O.; Ivanciuc, T.; Cabrol–Bass, D.; Balaban, A. T. Evaluation in Quantitative Structure–Property Relationship Models of Structural Descriptors Derived from Information–Theory Operators. J. Chem. Inf. Comput. Sci. 2000, 40, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Balaban, T.; Mills, D.; Ivanciuc, O.; Basak, S. C. Reverse Wiener Indices. Croat. Chem. Acta. 2000, 73, 923. [Google Scholar]
- Ivanciuc, O.; Ivanciuc, T.; Klein, D. J.; Seitz, W. A.; Balaban, A. T. Wiener Index Extension by Counting Even/Odd Graph Distances. J. Chem. Inf. Comput. Sci. 2001, 41, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Ivanciuc, O. Building–Block Computation of the Ivanciuc–Balaban Indices for the Virtual Screening of Combinatorial Libraries. Internet Electron. J. Mol. Des. 2002, 1, 1–9. http://www.biochempress.com. [Google Scholar]
- Rios–Santamarina, I.; García–Doménech, R.; Cortijo, J.; Santamaría, P.; Morcillo, E. J.; Gálvez, J. Natural Compounds with Bronchodilator Activity Selected by Molecular Topology. Internet Electron. J. Mol. Des. 2002, 1, 70–79. http://www.biochempress.com. [Google Scholar]
- Marino, D. J. G.; Peruzzo, P. J.; Castro, E. A.; Toropov, A. A. QSAR Carcinogenic Study of Methylated Polycyclic Aromatic Hydrocarbons Based on Topological Descriptors Derived from Distance Matrices and Correlation Weights of Local Graph Invariants. Internet Electron. J. Mol. Des. 2002, 1, 115–133. http://www.biochempress.com. [Google Scholar]
- Ivanciuc, O. QSAR Comparative Study of Wiener Descriptors for Weighted Molecular Graphs. J. Chem. Inf. Comput. Sci. 2000, 40, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E. Spectral Moment of Edge Adjacency Matrix in Molecular Graphs.1. Definition and Application to the Prediction of Physical Properties of Alkanes. J. Chem. Inf. Comp. Sci. 1996, 36, 846–849. [Google Scholar]
- Toropov, A. A.; Toropova, A. P. QSAR Modeling of Mutagenicity Based on Graphs of Atomic Orbitals. Internet Electron. J. Mol. Des. 2002, 1, 108–114. http://www.biochempress.com. [Google Scholar]
- Liu, S. S.; Liu, H. L.; Shi, Y. Y.; Wang, L. S. QSAR of Cyclooxygenase–2 (COX–2) Inhibition by 2,3-Diarylcyclopentenones Based on MEDV–13. Internet Electron. J. Mol. Des. 2002, 1, 310–318. http://www.biochempress.com. [Google Scholar]
- Estrada, E. The Structural Interpretation of the Randiæ Index. Internet Electron. J. Mol. Des. 2002, 1, 360–366. http://www.biochempress.com. [Google Scholar]
- Gute, B. D.; Basak, S. C.; Mills, D.; Hawkins, D. M. Tailored Similarity Spaces for the Prediction of Physicochemical Properties. Internet Electron. J. Mol. Des. 2002, 1, 374–387. http://www.biochempress.com. [Google Scholar]
- Lukovits, I.; Milièevic, A.; Nikolic, S.; Trinajstic, N. On Walk Counts and Complexity of General Graphs. Internet Electron. J. Mol. Des. 2002, 1, 388–400. http://www.biochempress.com. [Google Scholar]
- Cao, C.; Yuan, H. A. Modified Distance Matrix to Distinguish Cis/Trans Isomers of Cycloalkanes. Internet Electron. J. Mol. Des. 2002, 1, 401–409. http://www.biochempress.com. [Google Scholar]
- Marrero, Y.; Romero, V. TOMO-COMD sotfware. Central University of Las Villas, 2002. [Google Scholar]
- Cotton, F. A. Advanced Inorganic Chemistry; Ed. Revolucionaria: Cuba; p. 103.
- Randić, M. Generalized Molecular Descriptors. J. Math. Chem. 1991, 7, 155–168. [Google Scholar] [CrossRef]
- STATISTICA ver. 5.5, Statsoft, Inc., 1999.
- Belsey, D. A.; Kuh, E.; Welsch, R. E. Regression Diagnostics; Wiley: New York, 1980. [Google Scholar]
- Needham, D. E.; Chien, W. I.; Seybold, P. G. Molecular Modeling of the Physical Properties of the Alkanes. J. Am. Chem. Soc. 1998, 110, 4186–4194. [Google Scholar] [CrossRef]
- Alzina, R. B. Introduccion conceptual al análisis multivariable. Un enfoque informatico con los paquetes SPSS-X, BMDP, LISREL Y SPAD; PPU: SA Barcelona, 1989; charter 8; Vol. I, p. 202. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graphic Modell. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Conradi, R. A.; Burton, P. S.; Borchardt, R. T. Pliska, V., Testa, B., van de Waterbeemd, H., Eds.; Lipophilicity in Drug Action and Toxicology; VCH: Weinheim, 1996; pp. 233–252. [Google Scholar]
- Walters, W. P.; Stahl, M. T.; Murcko, M. A. Virtual Screening-an Overview. Drug Disc Today. 1998, 3, 160–178. [Google Scholar]
- Drie, J. H. V.; Lajinees, M. S. Approaches to Virtual Library Design. Drug Disc Today. 1998, 3, 274–283. [Google Scholar] [CrossRef]
- Julian-Ortiz, J. V.; Gálvez, J.; Muños-Collado, C.; García- Doménech, R.; Gimeno-Cardona, C. Virtual Combinatorial Synthesis and Computational Screening of New Potential Anti-Herpes Compounds. J Med Chem. 1999, 42, 3308–3314. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G. L.; Sinko, P. J.; Fleisher, D. Estimating Human Oral Fraction Dose Absorbed: A Correlation Using Rat Intestinal Membrane Permeability for Passive and Carrier-Mediated Compounds. Pharm Res. 1988, 5, 651–654. [Google Scholar]
- Chong, S.; Dando, S. A.; Morrison, R. Evaluation of Biocoat Intestinal Epithelium Differentiation Environment (Accelerated Cultured Caco-2 Cells) as an Absorption-Screening Model with Improved Productivity. Pharm Res. 1997, 14, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Rubas, W.; Jezyk, N.; Grass, G. M. Comparison of the Permeability Characteristics of a Human Colonic Epithelial (Caco-2) Cell Line to Colon of Rabbit, Monkey and Dog Intestine and Human Drug Absorption. Pharm Res. 1993, 10, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Yee, S. In vitro Permeability Across Caco-2 Cells (colonic) Can Predict in vivo (Small Intestinal) Absorption in Man-Fact or Myth. Pharm Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Grès, M.; Julian, B.; Bourrié, M.; Meunier, V.; Roques, C.; Berger, M.; Boulenc, X.; Berger, Y.; Fabre, G. Correlation Between Oral Drug Absorption in Humans and Apparent Drug Permeability in TC-7 Cells, a Human Epithelial Intestinal Cell Line: Comparison with the Parental Caco-2 Cell Line. Pharm Res. 1998, 15, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.; Janich, S.; Roessler, B.J.; Hilfinger, J. M.; Amidon, G. L. HT29-MTX/Caco-2 Cocultures as an in vitro Model for the Intestinal Epithelium: In vitro-in vivo Correlation with Permeability Data From Rats and Humans. J Pharm Sci. 1996, 85, 1070–1076. [Google Scholar]
- Stewar, B. H.; Chan, O. H.; Lu, R. H.; Reyner, E. L.; Schmid, H. L.; Hamilton, H. W.; Steinbaugh, B. A.; Taylor, M. D. Comparison of Intestinal Permeabilities Determined in Multiple in vitro and in situ Models: Relationships to Absorption in Humans. Pharm Res. 1995, 12, 693–699. [Google Scholar] [CrossRef] [PubMed]
© 2003 by MDPI (http://www.mdpi.org). Reproduction for noncommercial purposes permitted.
Share and Cite
Ponce, Y.M.; Pérez, M.A.C.; Zaldivar, V.R.; Ofori, E.; Montero, L.A. Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs. Int. J. Mol. Sci. 2003, 4, 512-536. https://doi.org/10.3390/i4080512
Ponce YM, Pérez MAC, Zaldivar VR, Ofori E, Montero LA. Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs. International Journal of Molecular Sciences. 2003; 4(8):512-536. https://doi.org/10.3390/i4080512
Chicago/Turabian StylePonce, Yovani Marrero, Miguel Angel Cabrera Pérez, Vicente Romero Zaldivar, Ernest Ofori, and Luis A. Montero. 2003. "Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs" International Journal of Molecular Sciences 4, no. 8: 512-536. https://doi.org/10.3390/i4080512
APA StylePonce, Y. M., Pérez, M. A. C., Zaldivar, V. R., Ofori, E., & Montero, L. A. (2003). Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs. International Journal of Molecular Sciences, 4(8), 512-536. https://doi.org/10.3390/i4080512