3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach

Abstract
:1. Introduction
2. Computational methods
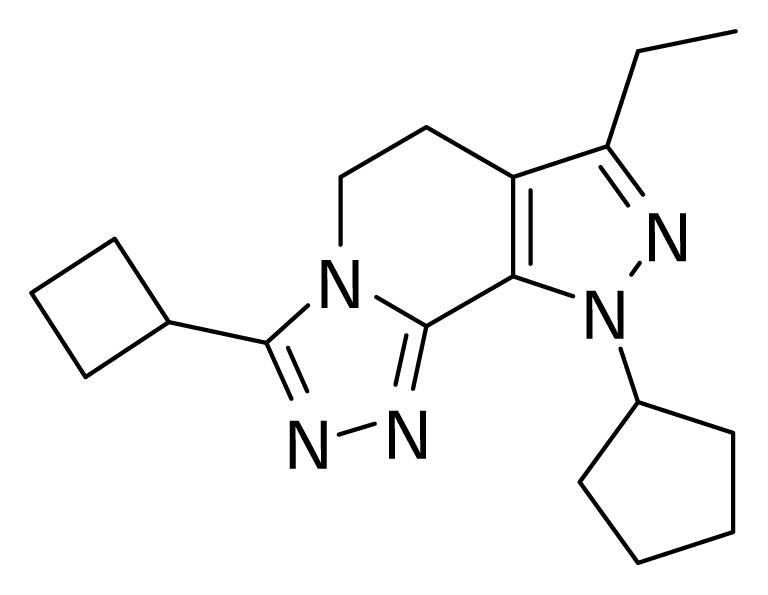
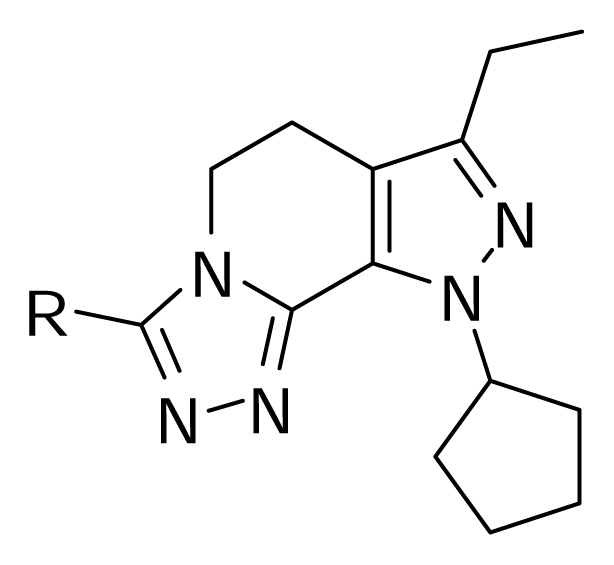
2.1 Molecular Modeling
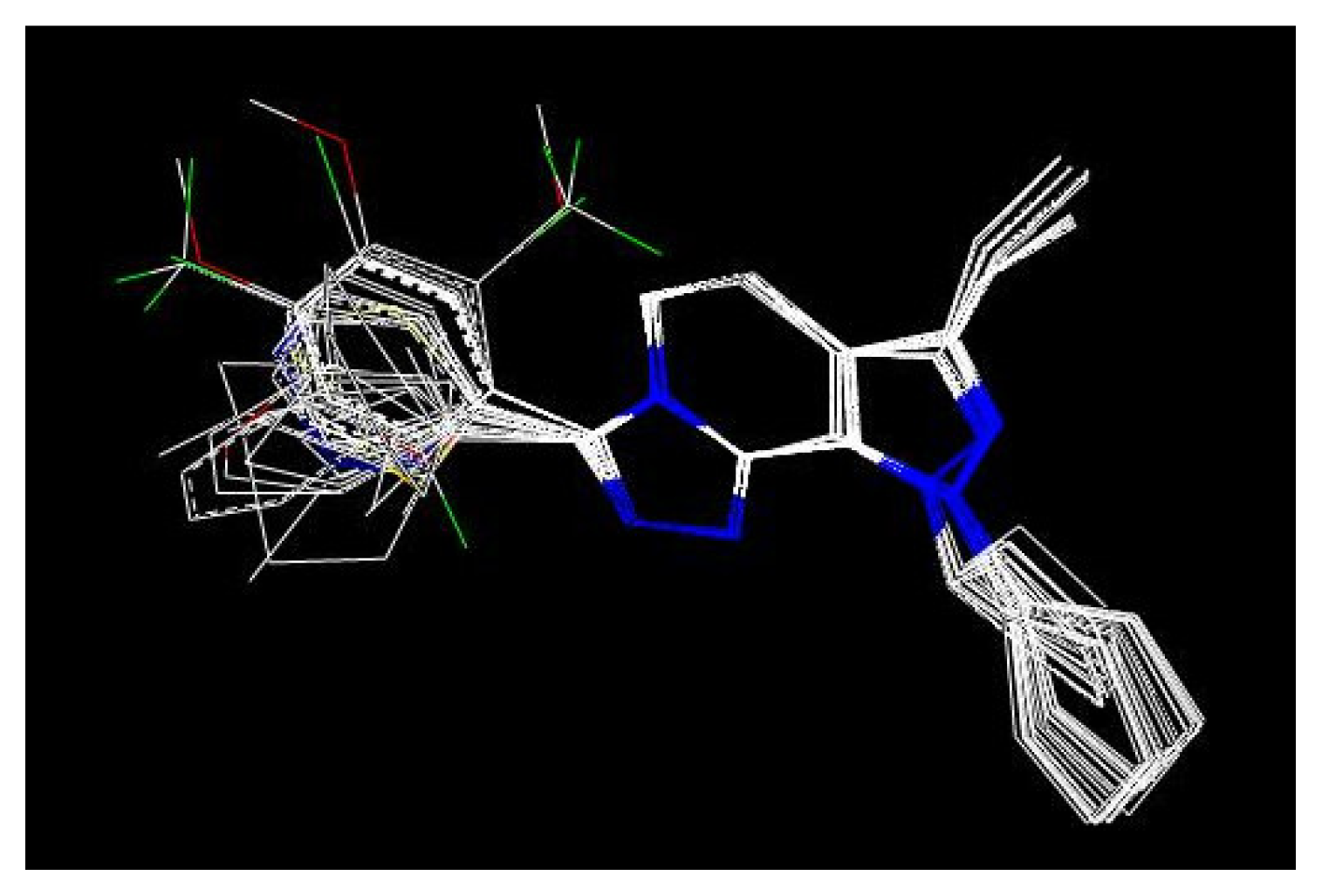
2.2 CoMFA analysis
2.3. Partial least squares (PLS) analysis
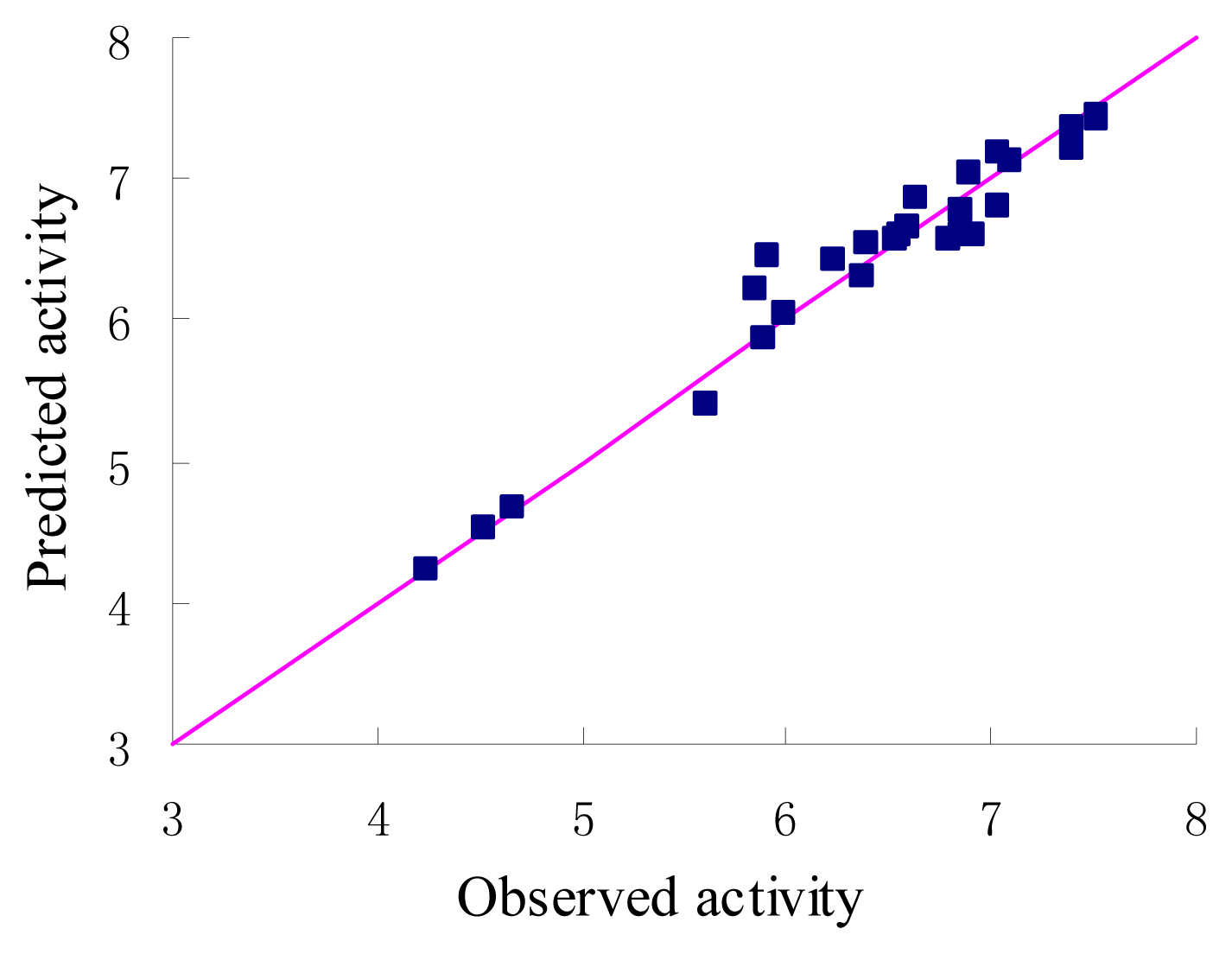
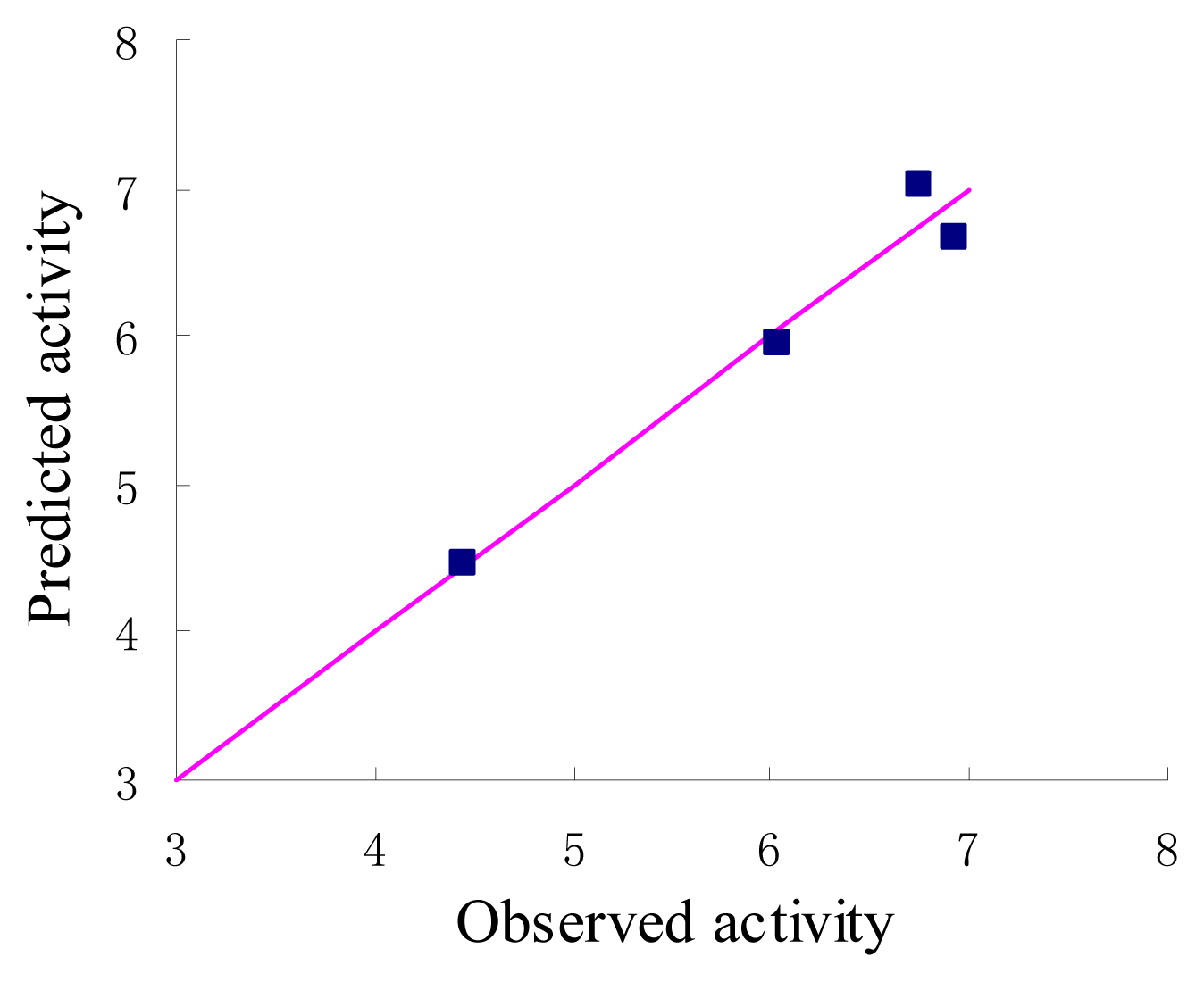
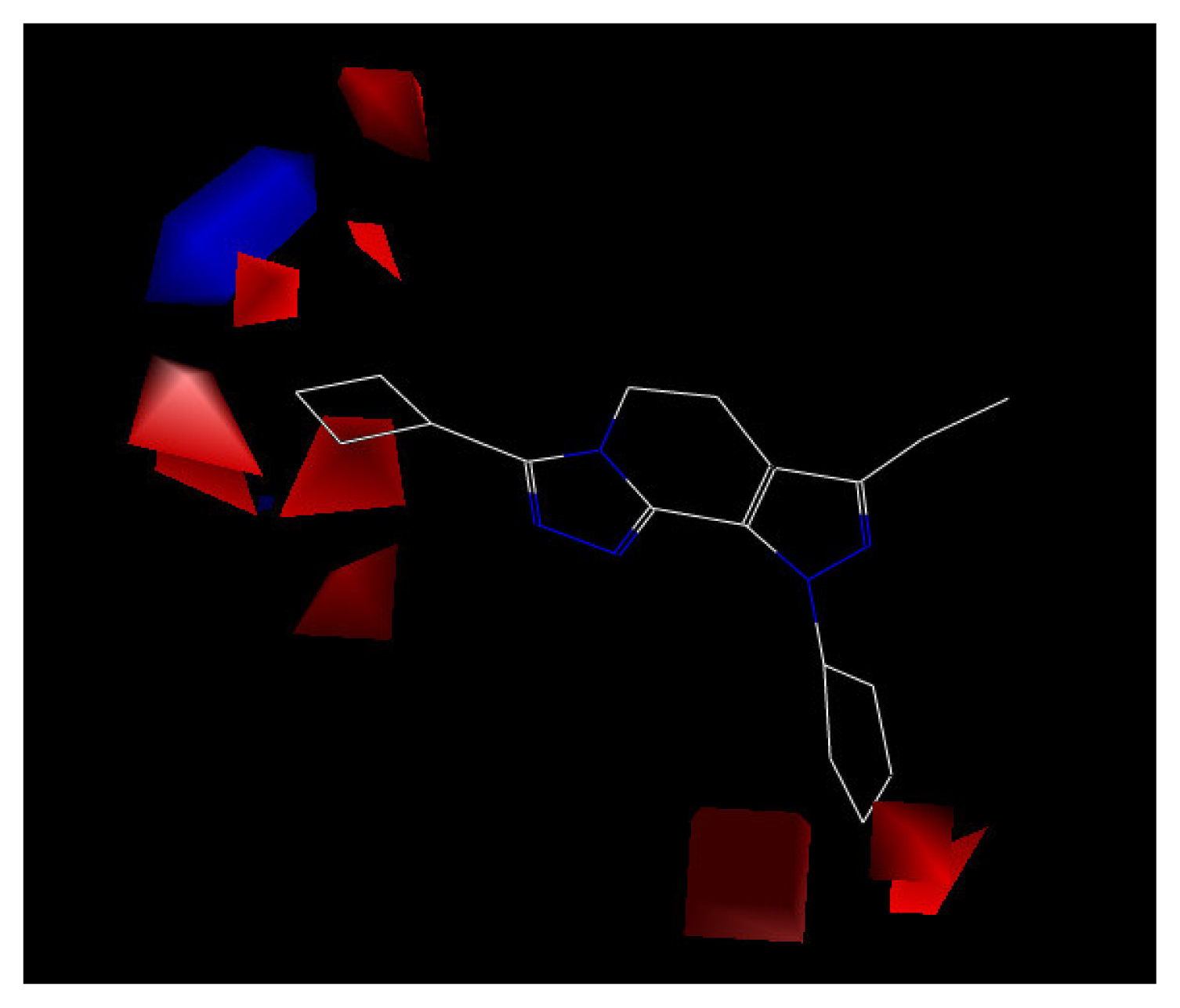
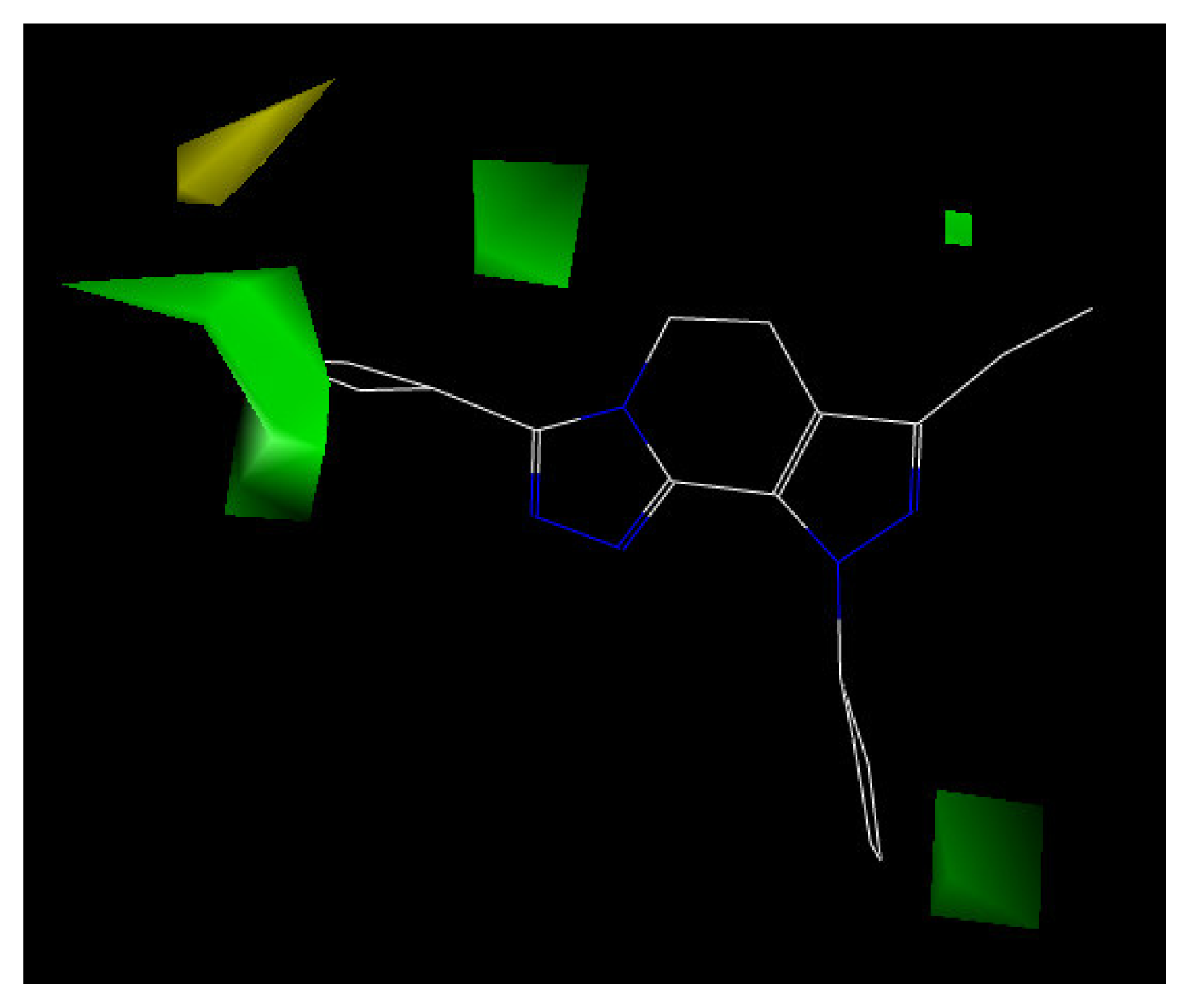
3. Results and Discussion
4. Conclusions







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | R | pIC50 (Observed) | pIC50 (Predicted) | Residual |
| 1△ | phenyl | 6.921 | 6.657 | 0.264 |
| 2 | 2-methoxyphenyl | 6.237 | 6.413 | −0.176 |
| 3 | 2-methylphenyl | 6.796 | 6.564 | 0.232 |
| 4 | 2-chlorophernyl | 6.538 | 6.559 | −0.021 |
| 5 | 2-trifluoromethylphenyl | 6.367 | 6.296 | 0.071 |
| 6 | 3-methoxyphenyl | 5.886 | 5.863 | 0.023 |
| 7 | 3-chlorophenyl | 5.854 | 6.223 | −0.369 |
| 8 | 4-methoxyphenyl | 4.523 | 4.527 | −0.004 |
| 9 | 4-methylphenyl | 4.658 | 4.664 | −0.006 |
| 10△ | 4-chlorophernyl | 4.444 | 4.471 | −0.027 |
| 11 | 4-trifluoromethylphenyl | 4.244 | 4.24 | 0.004 |
| 12 | 2-pyridyl | 6.585 | 6.659 | −0.074 |
| 13 | 3-pyridyl | 5.921 | 6.441 | −0.520 |
| 14 | 4-pyridyl | 7.046 | 6.812 | 0.234 |
| 15 | 2-furanyl | 6.553 | 6.582 | −0.029 |
| 16 | 2-thienyl | 6.854 | 6.591 | 0.263 |
| 17 | 3-chloro-4-methylthien-2-yl | 6.000 | 6.049 | −0.049 |
| 18 | benzyl | 6.398 | 6.551 | −0.153 |
| 19 | 3-thenyl | 6.921 | 6.595 | 0.326 |
| 20△ | methyl | 6.041 | 5.948 | 0.093 |
| 21 | ethyl | 6.886 | 7.021 | −0.135 |
| 22△ | propyl | 6.745 | 7.009 | −0.264 |
| 23 | butyl | 7.398 | 7.217 | 0.181 |
| 24 | cyclobutyl | 7.523 | 7.417 | 0.106 |
| 25 | cyclopentyl | 7.046 | 7.185 | −0.139 |
| 26 | cyclohexyl | 6.854 | 6.759 | 0.095 |
| 27 | 4-tetrahydropyranyl | 6.638 | 6.867 | −0.229 |
| 28 | 3-pentyl | 7.523 | 7.448 | 0.075 |
| 29 | 1-methylcyclohex-1-yl | 7.398 | 7.365 | 0.033 |
| 30 | tert-butyl | 7.097 | 7.132 | −0.035 |
| 31 | Bicycle[2.2.2]octanyl | 5.620 | 5.385 | 0.235 |
| grid spacing (Å) | 1.0 | 1.5 | 2.0 | 2.5 | 3.0 |
|---|---|---|---|---|---|
| q2 | 0.335 | 0.297 | 0.741 | 0.295 | 0.223 |
| r2 | 0.540 | 0.524 | 0.954 | 0.566 | 0.345 |
| standard error | 0.630 | 0.641 | 0.211 | 0.623 | 0.738 |
| PLS statistics | CoMFA |
|---|---|
| q2 (leave-one out cross-validated predicted power of model) | 0.741 |
| r2 (correlation coefficient squared of PLS analysis) | 0.954 |
| N (optimum number of components obtained from cross-validated PLS analysis and the same used in final non cross-validated analysis) | 5 |
| Standard error of estimate (SEE) | 0.211 |
| F-test value (F-value) | 103.397 |
| Steric field contribution from CoMFA | 0.915 |
| Electrostatic field contribution from CoMFA | 0.085 |
References and Notes
- Mallia, P.; Contoli, M.; Caramori, G.; Pandit, A.; Johnston, S.L.; Papi, A. Exacerbations of Asthma and Chronic Obstructive Pulmonary Disease (COPD): Focus on Virus Induced Exacerbations. Curr. Pharm. Des 2007, 13, 73–97. [Google Scholar]
- Liam, G.H.; John, T.L.; Lorcan, P.A.M. Inflammation in Chronic Obstructive Pulmonary Disease: Implications for New Treatment Strategies. Curr. Med. Chem 2007, 14, 187–796. [Google Scholar]
- Houslay, M.D.; Schafer, P.; Zhang, K.Y. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug. Discov. Today 2005, 10, 1503–1519. [Google Scholar]
- Huang, Z; Mancini, J.A. Phosphodiesterase 4 inhibitors for the treatment of asthma and COPD. Curr. Med. Chem. 2006, 13, 3253–3262. [Google Scholar]
- Barnes, P.J.; Stockley, R.A. COPD: current therapeutic interventions and future approaches. Eur. Respir. J 2005, 25, 1084–1106. [Google Scholar]
- Burnouf, C.; Pruniaux, M.P. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs. Curr. Pharm. Des 2002, 8, 1255–1296. [Google Scholar]
- Claus, k.; Martin, F. Phosphodiesterase-4 inhibitors as a novel approach for the treatment of respiratory disease: cilomilast. Expert. Opin. Investig. Drugs 2007, 16, 109–124. [Google Scholar]
- Duplantier, A.J.; Bachert, E.L.; Cheng, J.B.; Cohan, V.L.; Jenkinson, T.H.; Kraus, K.G.; Mckechney, M.W.; Pillar, J.D.; Watson, J.W. SAR of a Series of 5,6-Dihydro-(9H)-pyrazolo [3,4-c]-1,2,4-triazolo[4,3-α] pytidines as Potent Inhibitons of Human Eosinophil Phosphodiesterase. J. Med. Chem 2007, 50, 344–349. [Google Scholar]
- Lipworth, B.J. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 2005, 365, 167–175. [Google Scholar]
- Oding, J.O. Inhibitors of PDE4: a review of recent patent literature. Expert. Opin. Ther. Patents 2005, 15, 773–787. [Google Scholar]
- Huang, M.; Yang, D.Y.; Shang, Z.; Zou, J.; Yu, Q. 3D-QSAR Studies on 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors by Comparative Molecular Field Analysis (CoMFA). Bioorg. Med. Chem. Lett 2002, 12, 2271–2275. [Google Scholar]
- Ozlem, T.A.; Betul, T.G.; Yildiz, I.; Esin, A.S.; Ismail, Y. 3D-QSAR analysis on benzazole derivatives as eukaryotic topoisomerase II inhibitors by using comparative molecular field analysis method. Bioorg. Med. Chem 2005, 13, 6354–6359. [Google Scholar]
- Sybyl 7.0; Tripos Inc: St. Louis, USA.
- Hokuldsson, A. PLS regression methods. J. Chemometrics 1988, 2, 211–228. [Google Scholar]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc 1998, 110, 5959–5967. [Google Scholar]
- Xu, M.; Zhang, A.Q.; Han, S.K.; Wang, L.S. Studies of 3D-quantitative structure-activity relationships on a set of nitroaromatic compounds: CoMFA, advanced CoMFA and CoMSIA. Chemsphere 2002, 48, 707–715. [Google Scholar]
© 2007 by MDPI ( http://www.mdpi.org) Reproduction is permitted for noncommercial purposes.
Share and Cite
Yang, Z.; Sun, P. 3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach. Int. J. Mol. Sci. 2007, 8, 714-722. https://doi.org/10.3390/i8070714
Yang Z, Sun P. 3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach. International Journal of Molecular Sciences. 2007; 8(7):714-722. https://doi.org/10.3390/i8070714
Chicago/Turabian StyleYang, Zhaoqi, and Pinghua Sun. 2007. "3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach" International Journal of Molecular Sciences 8, no. 7: 714-722. https://doi.org/10.3390/i8070714
APA StyleYang, Z., & Sun, P. (2007). 3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach. International Journal of Molecular Sciences, 8(7), 714-722. https://doi.org/10.3390/i8070714