Thick and Thin Filament Gene Mutations in Striated Muscle Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
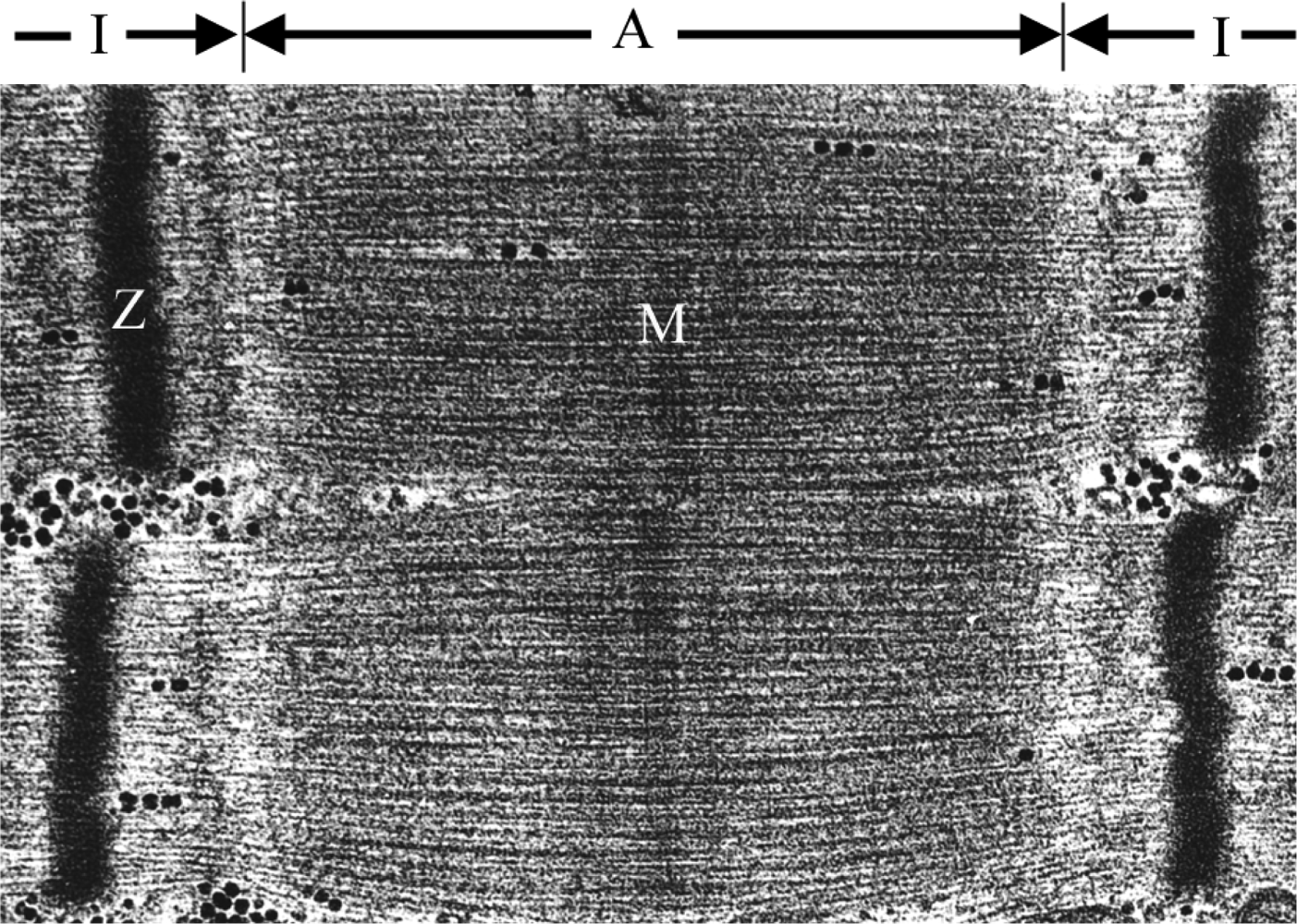
2. The Sarcomere
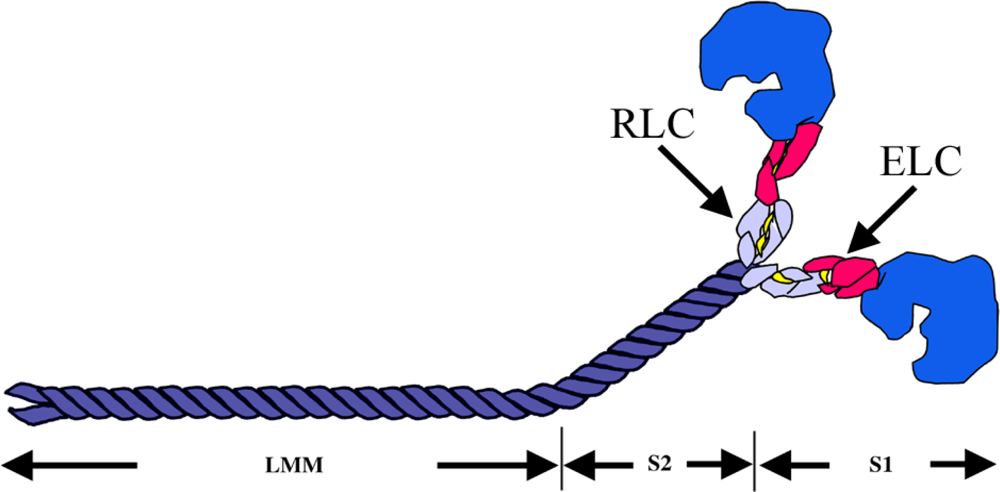
2.1. The Major Component of the Thick Filament: Myosin
2.2. The Major Components of the Thin Filament: Actin, Tropmyosin, Troponin Complex and Nebulin
3. Myopathies Involving Thick and Thin Filament Proteins
3.1. Disorders-associated Mutations Found in Thick Filament Components
3.1.1. Myosin Heavy Chains
3.1.1.1. MyHC IIa
3.1.1.2. Slow/β-cardiac MyHC
3.1.1.3. Perinatal MyHC
3.1.1.4. Embryonic MyHC
3.1.2. Myosin Light Chains
3.1.2.1. Regulatory Light Chain RLC
3.1.2.2. Essential Light Chain ELC
3.2. Disorder-associated Mutations Found in Thin Filament Components
3.2.1. Actin
3.2.2.α-TM
3.2.3. β-TM
3.2.4. γ-TM
3.2.5. Tn Complex
3.2.6. Nebulin
4. Conclusions
References
- Clark, KA; McElhinny, AS; Beckerle, MC; Gregorio, CC. Striated Muscle Cytoarchitecture: an Intricate Web of Form and Function. Annu Rev Cell Dev Biol 2002, 18, 637–706. [Google Scholar]
- Maruyama, K; Ebashi, S. Alpha-actinin, a New Structural Protein from Striated Muscle. II. Action on Actin. J Biochem 1965, 5, 13–19. [Google Scholar]
- Blanchard, A; Ohanian, V; Critchley, D. The Structure and Function of Alpha-actinin. J Muscle Res Cell Motil 1989, 10, 280–289. [Google Scholar]
- Ehler, E; Rothen, BM; Hammerle, SP; et al. Myofibrillogenesis in the Developing Chicken Heart: Assembly of Z-Disk, M-Line and the Thick Filaments. J Cell Sci 1999, 112, 1529–1539. [Google Scholar]
- Lazarides, E. Desmin and Intermediate Filaments in Muscle Cells. Results Probl Cell Differ 1980, 11, 124–131. [Google Scholar]
- Koretz, JF. Effects of C-protein on Synthetic Myosin Filament Structure. Biophys J 1979, 27, 433–446. [Google Scholar]
- Davis, JS. Interaction of C-protein with pH 8.0 Synthetic Thick Filaments Prepared from the Myosin of Vertebrate Skeletal Muscle. J Muscle Res Cell Motil 1988, 9, 174–183. [Google Scholar]
- Winegrad, S. Cardiac Myosin Binding Protein C. Circ Res 1999, 84, 1117–1126. [Google Scholar]
- Spudich, JA. How Molecular Motors Work. Nature 1994, 372, 515–518. [Google Scholar]
- Ruppel, KM; Spudich, JA. Structure-Function Analysis of the Motor Domain of Myosin. Annu Rev Cell Dev Biol 1996, 12, 543–573. [Google Scholar]
- Rayment, I; Holden, HM; Whittaker, M; et al. Structure of the Actin-Myosin Complex and Its Implications for Muscle Contraction. Science 1993, 261, 58–65. [Google Scholar]
- Chen, T; Reisler, E. Tryptic Digestion of Rabbit Skeletal Myofibrils: An Enzymatic Probe of Myosin Cross-Bridges. Biochemistry 1984, 23, 2400–2407. [Google Scholar]
- Assulin, O; Werber, MM; Muhlrad, A. Effect of the Integrity of the Myofibrillar Structure on the Tryptic Accessibility of a Hinge Region of the Myosin Rod. FEBS Lett 1986, 197, 328–334. [Google Scholar]
- Weiss, A; Schiaffino, S; Leinwand, LA. Comparative Sequence Analysis of the Complete Human Sarcomeric Myosin Heavy Chain Family: Implications for Functional Diversity. J Mol Biol 1999, 290, 61–75. [Google Scholar]
- Rayment, I; Rypniewski, WR; Schmidt-Base, K; et al. Three-Dimensional Structure of Myosin Subfragment-1: A Molecular Motor. Science 1993, 261, 50–58. [Google Scholar]
- Weiss, A; Leinwand, LA. The Mammalian Myosin Heavy Chain Gene Family. Annu Rev Cell Dev Biol 1996, 12, 417–439. [Google Scholar]
- Leinwand, LA; Saez, L; McNally, E; Nadal-Ginard, B. Isolation and Characterization of Human Myosin Heavy Chain Genes. Proc Natl Acad Sci USA 1983, 80, 3716–3720. [Google Scholar]
- Weiss, A; Mayer, DC; Leinwand, LA. Diversity of Myosin-based Motility: Multiple Genes and Functions. Soc Gen Physiol Ser 1994, 49, 159–171. [Google Scholar]
- Saez, LJ; Gianola, KM; McNally, EM; et al. Human Cardiac Myosin Heavy Chain Genes and Their Linkage in the Genome. Nucleic Acids Res 1987, 15, 5443–5459. [Google Scholar]
- Smerdu, V; Karsch-Mizrachi, I; Campione, M; et al. Type IIx Myosin Heavy Chain Transcripts are Expressed in Type IIb Fibers of Human Skeletal Muscle. Am J Physiol 1994, 267, C1723–1728. [Google Scholar]
- Pedrosa-Domellof, F; Holmgren, Y; Lucas, CA; et al. Human Extraocular Muscles: Unique Pattern of Myosin Heavy Chain Expression During Myotube Formation. Invest Ophthalmol Vis Sci 2000, 41, 1608–1616. [Google Scholar]
- Sartore, S; Mascarello, F; Rowlerson, A; et al. Fibre Types in Extraocular Muscles: A New Myosin Isoform in the Fast Fibres. J Muscle Res Cell Motil 1987, 8, 161–172. [Google Scholar]
- Asmussen, G; Traub, I; Pette, D. Electrophoretic Analysis of Myosin Heavy Chain Isoform Patterns in Extraocular Muscles of the Rat. FEBS Lett 1993, 335, 243–245. [Google Scholar]
- Wieczorek, DF; Periasamy, M; Butler-Browne, GS; et al. Co-expression of Multiple Myosin Heavy Chain Genes, in Addition to a Tissue-Specific One, in Extraocular Musculature. J Cell Biol 1985, 101, 618–629. [Google Scholar]
- Gordon, AM; Homsher, E; Regnier, M. Regulation of Contraction in Striated Muscle. Physiol Rev 2000, 80, 853–924. [Google Scholar]
- Perry, SV. Vertebrate Tropomyosin: Distribution, Properties and Function. J Muscle Res Cell Motil 2001, 22, 5–49. [Google Scholar]
- Pieples, K; Wieczorek, DF. Tropomyosin 3 Increases Striated Muscle Isoform Diversity. Biochemistry 2000, 39, 8291–8297. [Google Scholar]
- Gahlmann, R; Wade, R; Gunning, P; Kedes, L. Differential Expression of Slow and Fast Skeletal Muscle Troponin C. Slow Skeletal Muscle Troponin C is Expressed in Human Fibroblasts. J Mol Biol 1988, 201, 379–391. [Google Scholar]
- Schreier, T; Kedes, L; Gahlmann, R. Cloning, Structural Analysis, and Expression of the Human Slow Twitch Skeletal Muscle/Cardiac Troponin C Gene. J Biol Chem 1990, 265, 21247–21253. [Google Scholar]
- Syska, H; Perry, SV; Trayer, IP. A New Method of Preparation of Troponin I (Inhibitory Protein) Using Affinity Chromatography. Evidence for Three Different Forms of Troponin I in Striated Muscle. FEBS Lett 1974, 40, 253–257. [Google Scholar]
- Barton, PJ; Cullen, ME; Townsend, PJ; et al. Close Physical Linkage of Human Troponin Genes: Organization, Sequence, and Expression of the Locus Encoding Cardiac Troponin I and Slow Skeletal Troponin T. Genomics 1999, 57, 102–109. [Google Scholar]
- Huang, P; Jin, X; Chen, Y; et al. Use of a Mixed-Mode Packing and Voltage Tuning for Peptide Mixture Separation in Pressurized Capillary Electrochromatography with an Ion Trap Storage/Reflectron Time-of-Flight Mass Spectrometer Detector. Anal Chem 1999, 71, 1786–1791. [Google Scholar]
- Wang, K. Titin/Connectin and Nebulin: Giant Protein Rulers of Muscle Structure and Function. Adv Biophys 1996, 33, 123–134. [Google Scholar]
- Horowits, R; Luo, G; Zhang, JQ; Herrera, AH. Nebulin and Nebulin-Related Proteins in Striated Muscle. Adv Biophys 1996, 33, 143–150. [Google Scholar]
- McElhinny, AS; Schwach, C; Valichnac, M; et al. Nebulin Regulates the Assembly and Lengths of the Thin Filaments in Striated Muscle. J Cell Biol 2005, 170, 947–957. [Google Scholar]
- Geisterfer-Lowrance, AA; Kass, S; Tanigawa, G; et al. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A Beta Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006. [Google Scholar]
- Martinsson, T; Oldfors, A; Darin, N; et al. Autosomal Dominant Myopathy: Missense Mutation (Glu-706 → Lys) in the Myosin Heavy Chain IIa Gene. Proc Natl Acad Sci USA 2000, 97, 14614–14619. [Google Scholar]
- Laing, NG; Laing, BA; Meredith, C; et al. Autosomal Dominant Distal Myopathy: Linkage to Chromosome 14. Am J Hum Genet 1995, 56, 422–427. [Google Scholar]
- Meredith, C; Herrmann, R; Parry, C; et al. Mutations in the Slow Skeletal Muscle Fiber Myosin Heavy Chain Gene (MYH7) Cause Laing Early-Onset Distal Myopathy (MPD1). Am J Hum Genet 2004, 75, 703–708. [Google Scholar]
- Lamont, PJ; Udd, B; Mastaglia, FL; et al. Laing Early Onset Distal Myopathy: Slow Myosin Defect with Variable Abnormalities on Muscle Biopsy. J Neurol Neurosurg Psychiatry 2006, 77, 208–215. [Google Scholar]
- Scoppetta, C; Casali, C; La Cesa, I; et al. Infantile Autosomal Dominant Distal Myopathy. Acta Neurol Scand 1995, 92, 122–126. [Google Scholar]
- Tajsharghi, H; Thornell, LE; Lindberg, C; et al. Myosin Storage Myopathy Associated with a Heterozygous Missense Mutation in MYH7. Ann Neurol 2003, 54, 494–500. [Google Scholar]
- Bohlega, S; Abu-Amero, SN; Wakil, SM; et al. Mutation of the Slow Myosin Heavy Chain Rod Domain Underlies Hyaline Body Myopathy. Neurology 2004, 62, 1518–1521. [Google Scholar]
- Laing, NG; Ceuterick-de Groote, C; Dye, DE; et al. Myosin Storage Myopathy: Slow Skeletal Myosin (MYH7) Mutation in Two Isolated Cases. Neurology 2005, 64, 527–529. [Google Scholar]
- Shingde, MV; Spring, PJ; Maxwell, A; et al. Myosin Storage (Hyaline Body) Myopathy: A Case Report. Neuromuscul Disord 2006, 16, 882–886. [Google Scholar]
- Dye, DE; Azzarelli, B; Goebel, HH; Laing, NG. Novel Slow-Skeletal Myosin (MYH7) Mutation in the Original Myosin Storage Myopathy Kindred. Neuromuscul Disord 2006, 16, 357–360. [Google Scholar]
- Veugelers, M; Bressan, M; McDermott, DA; et al. Mutation of Perinatal Myosin Heavy Chain Associated with a Carney Complex Variant. N Engl J Med 2004, 351, 460–469. [Google Scholar]
- Toydemir, RM; Chen, H; Proud, VK; et al. Trismus-pseudocamptodactyly Syndrome is Caused by Recurrent Mutation of MYH8. Am J Med Genet 2006, 140, 2387–2393. [Google Scholar]
- Toydemir, RM; Rutherford, A; Whitby, FG; et al. Mutations in Embryonic Myosin Heavy Chain (MYH3) Cause Freeman-Sheldon Syndrome and Sheldon-Hall Syndrome. Nat Genet 2006, 38, 561–565. [Google Scholar]
- Darin, N; Kyllerman, M; Wahlstrom, J; et al. Autosomal Dominant Myopathy with Congenital Joint Contractures, Ophthalmoplegia, and Rimmed Vacuoles. Ann Neurol 1998, 44, 242–248. [Google Scholar]
- Tajsharghi, H; Darin, N; Rekabdar, E; et al. Mutations and Sequence Variation in the Human Myosin Heavy Chain IIa Gene (MYH2). Eur J Hum Genet 2005, 13, 617–622. [Google Scholar]
- Watkins, H; Rosenzweig, A; Hwang, DS; et al. Characteristics and Prognostic Implications of Myosin Missense Mutations in Familial Hypertrophic Cardiomyopathy. N Engl J Med 1992, 326, 1108–1114. [Google Scholar]
- Seidman, JG; Seidman, C. The Genetic Basis for Cardiomyopathy: from Mutation Identification to Mechanistic Paradigms. Cell 2001, 104, 557–567. [Google Scholar]
- Fananapazir, L; Dalakas, MC; Cyran, F; et al. Missense Mutations in the Beta-Myosin Heavy-Chain Gene Cause Central Core Disease in Hypertrophic Cardiomyopathy. Proc Natl Acad Sci USA 1993, 90, 3993–3997. [Google Scholar]
- Cuda, G; Fananapazir, L; Epstein, ND; Sellers, JR. The in vitro Motility Activity of Beta-Cardiac Myosin Depends on the Nature of the Beta-Myosin Heavy Chain Gene Mutation in Hypertrophic Cardiomyopathy. J Muscle Res Cell Motil 1997, 18, 275–283. [Google Scholar]
- Darin, N; Tajsharghi, H; Ostman-Smith, I; et al. New Skeletal Myopathy and Cardiomyopathy Associated with a Missense Mutation in MYH7. Neurology 2007, 68, 2041–2042. [Google Scholar]
- Overeem, S; Schelhaas, HJ; Blijham, PJ; et al. Symptomatic Distal Myopathy with Cardiomyopathy due to a MYH7 Mutation. Neuromuscul Disord 2007, 17, 490–493. [Google Scholar]
- Cancilla, PA; Kalyanaraman, K; Verity, MA; et al. Familial Myopathy with Probable Lysis of Myofibrils in Type I Fibers. Neurology 1971, 21, 579–585. [Google Scholar]
- Barohn, RJ; Brumback, RA; Mendell, JR; et al. Hyaline Body Myopathy. Neuromuscul Disord 1994, 4, 257–262. [Google Scholar]
- Masuzugawa, S; Kuzuhara, S; Narita, Y; et al. Autosomal Dominant Hyaline Body Myopathy Presenting as Scapuloperoneal Syndrome: Clinical Features and Muscle Pathology. Neurology 1997, 48, 253–257. [Google Scholar]
- Pegoraro, E; Gavassini, BF; Borsato, C; et al. MYH7 Gene Mutation in Myosin Storage Myopathy and Scapulo-Peroneal Myopathy. Neuromuscul Disord 2007, 17, 321–329. [Google Scholar]
- Hall, JG; Reed, SD; Greene, G. The Distal Arthrogryposes: Delineation of New Entities-Review and Nosologic Discussion. Am J Med Genet 1982, 11, 185–239. [Google Scholar]
- Bamshad, M; Jorde, LB; Carey, JC. A Revised and Extended Classification of the Distal Arthrogryposes. Am J Med Genet 1996, 65, 277–281. [Google Scholar]
- Tajsharghi, H; Kimber, E; Kroksmark, AK; et al. Embryonic Myosin Heavy Chain Mutations Cause Distal Arthrogryposis and Developmental Myosin Myopathy That Persists Postnatally. Arch Neurol. in press.
- Andersen, PS; Havndrup, O; Bundgaard, H; et al. Myosin Light Chain Mutations in Familial Hypertrophic Cardiomyopathy: Phenotypic Presentation and Frequency in Danish and South African Populations. J Med Genet 2001, 38, E43. [Google Scholar]
- Flavigny, J; Richard, P; Isnard, R; et al. Identification of Two Novel Mutations in the Ventricular Regulatory Myosin Light Chain Gene (MYL2) Associated with Familial and Classical Forms of Hypertrophic Cardiomyopathy. J Mol Med 1998, 76, 208–214. [Google Scholar]
- Poetter, K; Jiang, H; Hassanzadeh, S; et al. Mutations in Either the Essential or Regulatory Light Chains of Myosin are Associated with a Rare Myopathy in Human Heart and Skeletal Muscle. Nat Genet 1996, 13, 63–69. [Google Scholar]
- Richard, P; Charron, P; Carrier, L; et al. Hypertrophic Cardiomyopathy: Distribution of Disease Genes, Spectrum of Mutations, and Implications for a Molecular Diagnosis Strategy. Circulation 2003, 107, 2227–2232. [Google Scholar]
- Kabaeva, ZT; Perrot, A; Wolter, B; et al. Systematic Analysis of the Regulatory and Essential Myosin Light Chain Genes: Genetic Variants and Mutations in Hypertrophic Cardiomyopathy. Eur J Hum Genet 2002, 10, 741–748. [Google Scholar]
- Hernandez, OM; Jones, M; Guzman, G; et al. Myosin Essential Light Chain in Health and Disease. Am J Physiol 2007, 292, H1643–1654. [Google Scholar]
- Morano, I. Tuning the Human Heart Molecular Motors by Myosin Light Chains. J Mol Med 1999, 77, 544–555. [Google Scholar]
- Nowak, KJ; Wattanasirichaigoon, D; Goebel, HH; et al. Mutations in the Skeletal Muscle Alpha-Actin Gene in Patients with Actin Myopathy and Nemaline Myopathy. Nat Genet 1999, 23, 208–212. [Google Scholar]
- Laing, NG; Clarke, NF; Dye, DE; et al. Actin Mutations are One Cause of Congenital Fibre Type Disproportion. Ann Neurol 2004, 56, 689–694. [Google Scholar]
- Kaindl, AM; Ruschendorf, F; Krause, S; et al. Missense Mutations of ACTA1 Cause Dominant Congenital Myopathy with Cores. J Med Genet 2004, 41, 842–848. [Google Scholar]
- Donner, K; Ollikainen, M; Ridanpaa, M; et al. Mutations in the Beta-Tropomyosin (TPM2) Gene-a Rare Cause of Nemaline Myopathy. Neuromuscul Disord 2002, 12, 151–158. [Google Scholar]
- Tajsharghi, H; Ohlsson, M; Lindberg, C; et al. Congenital Myopathy with Nemaline Rods and Cap Structures Caused by a Mutation in the Beta-Tropomyosin Gene (TPM2). Arch Neurol 2007, 64, 1334–1338. [Google Scholar]
- Lehtokari, VL; Ceuterick-de Groote, C; de Jonghe, P; et al. Cap Disease Caused by Heterozygous Deletion of the Beta-Tropomyosin Gene TPM2. Neuromuscul Disord 2007, 17, 433–442. [Google Scholar]
- Sung, SS; Brassington, AM; Grannatt, K; et al. Mutations in Genes Encoding Fast-Twitch Contractile Proteins Cause Distal Arthrogryposis Syndromes. Am J Hum Genet 2003, 72, 681–690. [Google Scholar]
- Tajsharghi, H; Kimber, E; Holmgren, D; et al. Distal Arthrogryposis and Muscle Weakness Associated with a Beta-Tropomyosin Mutation. Neurology 2007, 68, 772–775. [Google Scholar]
- Laing, NG; Wilton, SD; Akkari, PA; et al. A Mutation in the Alpha Tropomyosin Gene TPM3 Associated with Autosomal Dominant Nemaline Myopathy. Nat Genet 1995, 9, 75–79. [Google Scholar]
- Wattanasirichaigoon, D; Swoboda, KJ; Takada, F; et al. Mutations of the Slow Muscle Alpha-Tropomyosin Gene, TPM3, Are a Rare Cause of Nemaline Myopathy. Neurology 2002, 59, 613–617. [Google Scholar]
- Tan, P; Briner, J; Boltshauser, E; et al. Homozygosity for a Nonsense Mutation in the Alpha-Tropomyosin Slow Gene TPM3 in a Patient with Severe Infantile Nemaline Myopathy. Neuromuscul Disord 1999, 9, 573–579. [Google Scholar]
- Durling, HJ; Reilich, P; Muller-Hocker, J; et al. De Novo Missense Mutation in a Constitutively Expressed Exon of the Slow Alpha-Tropomyosin Gene TPM3 Associated with an Atypical, Sporadic Case of Nemaline Myopathy. Neuromuscul Disord 2002, 12, 947–951. [Google Scholar]
- Penisson-Besnier, I; Monnier, N; Toutain, A; et al. A Second Pedigree with Autosomal Dominant Nemaline Myopathy Caused by TPM3 Mutation: A Clinical and Pathological Study. Neuromuscul Disord 2007, 17, 330–337. [Google Scholar]
- Clarke, NF; Kolski, H; Dye, DE; et al. Mutations in TPM3 Are a Common Cause of Congenital Fiber Type Disproportion. Ann Neurol 2008, 63, 329–337. [Google Scholar]
- Shrimpton, AE; Hoo, JJ. A TNNI2 Mutation in a Family with Distal Arthrogryposis Type 2B. European journal of medical genetics 2006, 49, 201–206. [Google Scholar]
- Kimber, E; Tajsharghi, H; Kroksmark, AK; et al. A Mutation in the Fast Skeletal Muscle Troponin I Gene Causes Myopathy and Distal Arthrogryposis. Neurology 2006, 67, 597–601. [Google Scholar]
- Jiang, M; Zhao, X; Han, W; et al. A Novel Deletion in TNNI2 Causes Distal Arthrogryposis in a Large Chinese Family with Marked Variability of Expression. Hum Genet 2006, 120, 238–242. [Google Scholar]
- Johnston, JJ; Kelley, RI; Crawford, TO; et al. A Novel Nemaline Myopathy in the Amish Caused by a Mutation in Troponin T1. Am J Hum Genet 2000, 67, 814–821. [Google Scholar]
- Sung, SS; Brassington, AM; Krakowiak, PA; et al. Mutations in TNNT3 Cause Multiple Congenital Contractures: A Second Locus for Distal Arthrogryposis Type 2B. Am J Hum Genet 2003, 73, 212–214. [Google Scholar]
- Hoffmann, B; Schmidt-Traub, H; Perrot, A; et al. First Mutation in Cardiac Troponin C, L29Q, in a Patient with Hypertrophic Cardiomyopathy. Hum Mutat 2001, 17, 524. [Google Scholar]
- Mogensen, J; Murphy, RT; Shaw, T; et al. Severe Disease Expression of Cardiac Troponin C and T Mutations in Patients with Idiopathic Dilated Cardiomyopathy. J Am Coll Cardiol 2004, 44, 2033–2040. [Google Scholar]
- Pelin, K; Hilpela, P; Donner, K; et al. Mutations in the Nebulin Gene Associated with Autosomal Recessive Nemaline Myopathy. Proc Natl Acad Sci USA 1999, 96, 2305–2310. [Google Scholar]
- Pelin, K; Donner, K; Holmberg, M; et al. Nebulin Mutations in Autosomal Recessive Nemaline Myopathy: An Update. Neuromuscul Disord 2002, 12, 680–686. [Google Scholar]
- Wallgren-Pettersson, C; Donner, K; Sewry, C; et al. Mutations in the Nebulin Gene Can Cause Severe Congenital Nemaline Myopathy. Neuromuscul Disord 2002, 12, 674–679. [Google Scholar]
- Lehtokari, VL; Pelin, K; Sandbacka, M; et al. Identification of 45 Novel Mutations in the Nebulin Gene Associated with Autosomal Recessive Nemaline Myopathy. Hum Mutat 2006, 27, 946–956. [Google Scholar]
- Wallgren-Pettersson, C; Lehtokari, VL; Kalimo, H; et al. Distal Myopathy Caused by Homozygous Missense Mutations in the Nebulin Gene. Brain 2007, 130, 1465–1476. [Google Scholar]





Share and Cite
Tajsharghi, H. Thick and Thin Filament Gene Mutations in Striated Muscle Diseases. Int. J. Mol. Sci. 2008, 9, 1259-1275. https://doi.org/10.3390/ijms9071259
Tajsharghi H. Thick and Thin Filament Gene Mutations in Striated Muscle Diseases. International Journal of Molecular Sciences. 2008; 9(7):1259-1275. https://doi.org/10.3390/ijms9071259
Chicago/Turabian StyleTajsharghi, Homa. 2008. "Thick and Thin Filament Gene Mutations in Striated Muscle Diseases" International Journal of Molecular Sciences 9, no. 7: 1259-1275. https://doi.org/10.3390/ijms9071259
APA StyleTajsharghi, H. (2008). Thick and Thin Filament Gene Mutations in Striated Muscle Diseases. International Journal of Molecular Sciences, 9(7), 1259-1275. https://doi.org/10.3390/ijms9071259