
2-Hydroxy-5-(3,5,7-trihydroxy-4-oxo-4H-chromen-2-yl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate: Synthesis, In Silico Analysis and In Vitro Pharmacological Evaluation

 ,
,  ,
,  and
and
Abstract
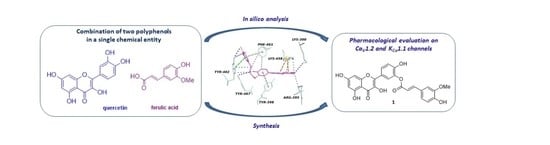
:
1. Introduction
2. Results and Discussion
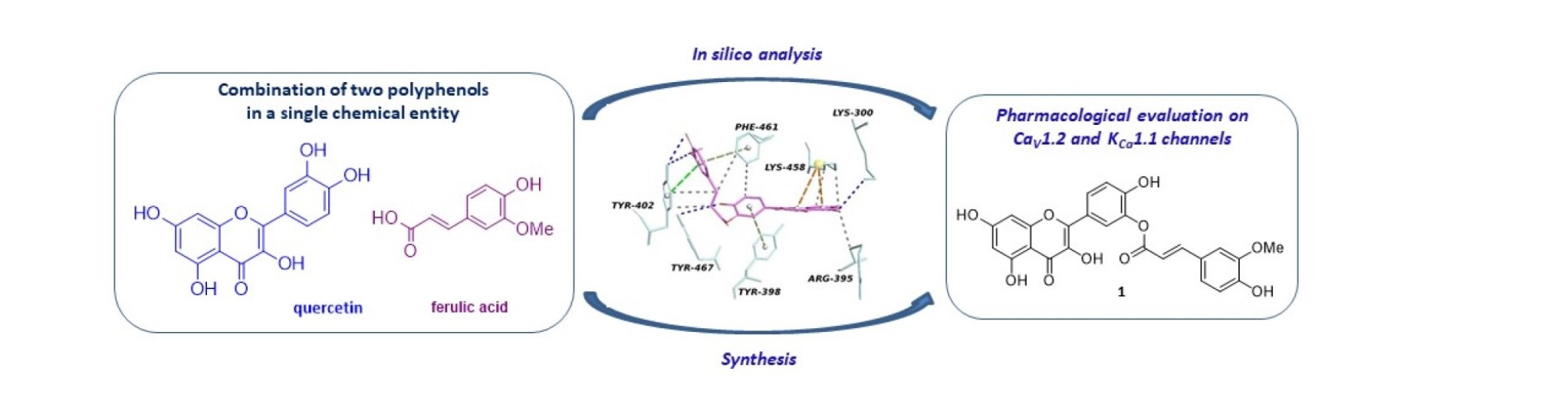
2.1. In Silico Analysis
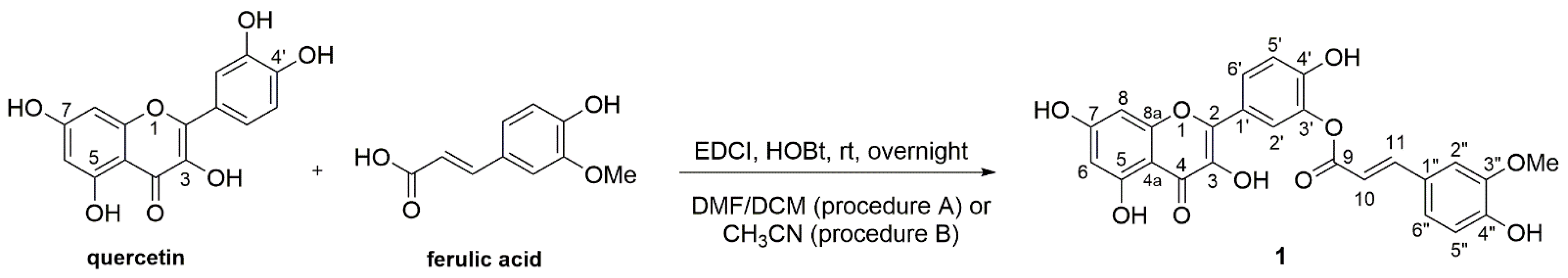
2.2. Chemistry
2.3. Pharmacological Evaluation
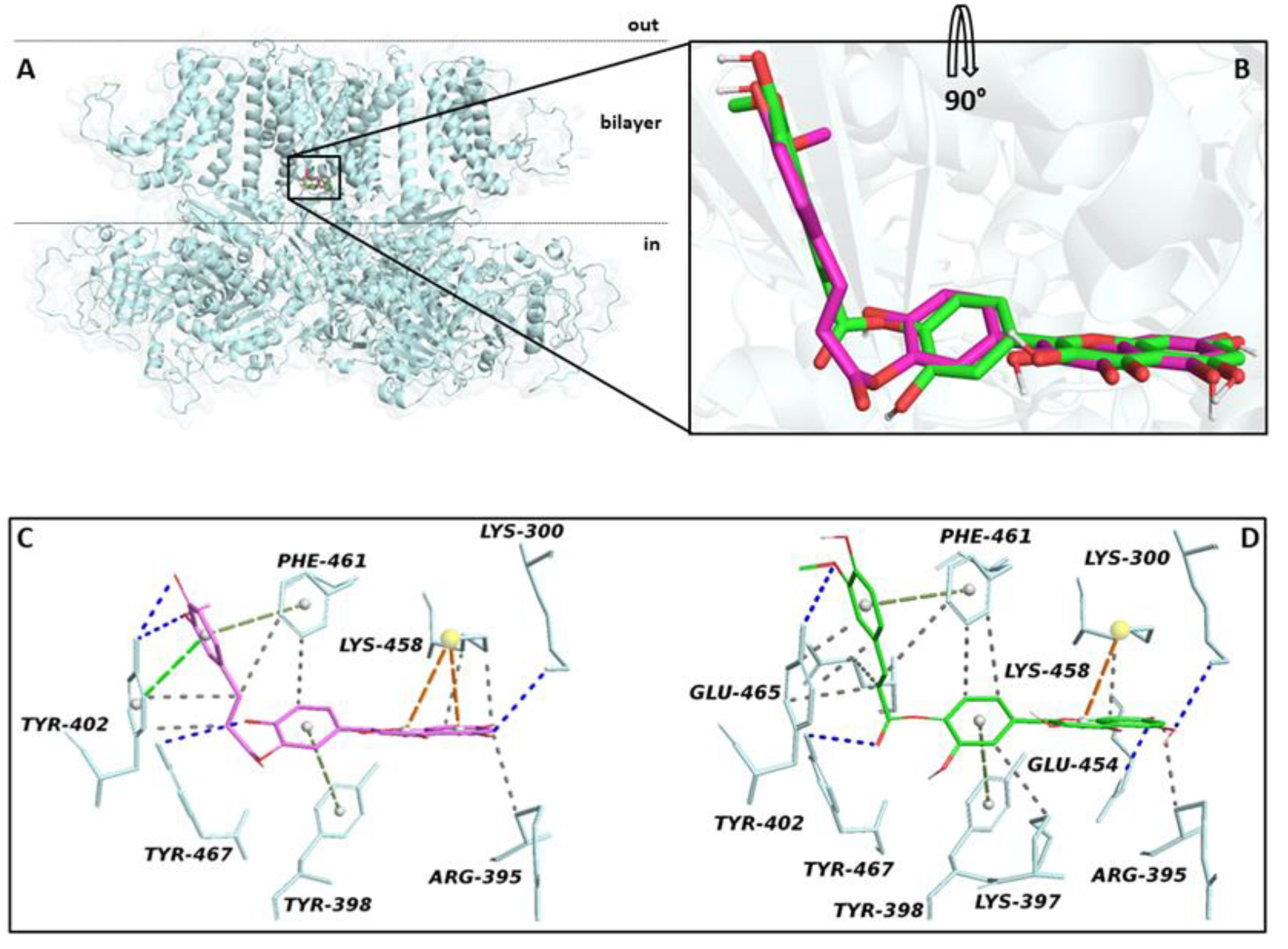
2.3.1. Effect of Compound 1 on IKCa1.1
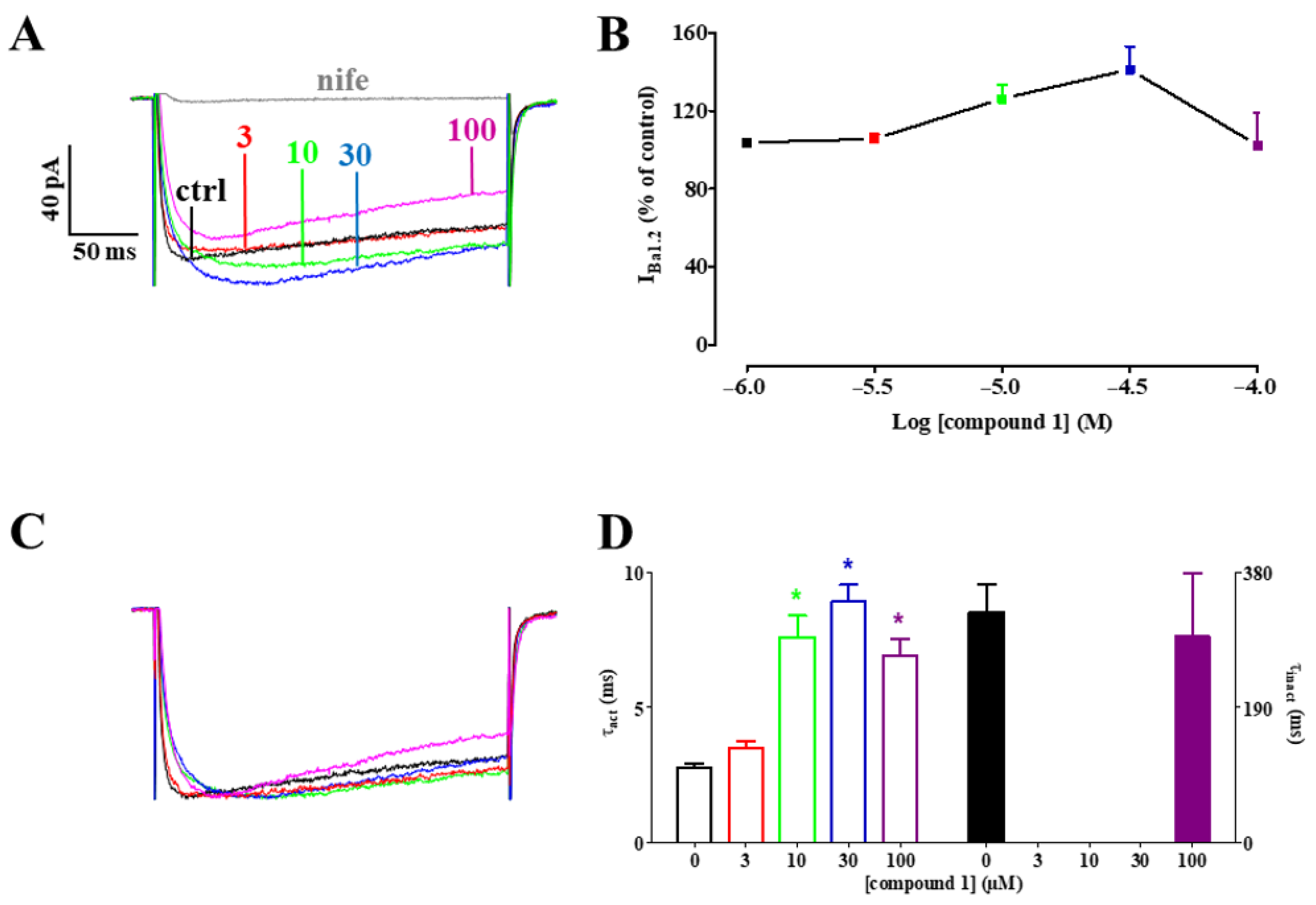
2.3.2. Effect of Compound 1 on IBa1.2
3. Materials and Methods
3.1. In Silico Methods
3.2. Chemistry
3.2.1. General
3.2.2. Synthesis of 2-Hydroxy-5-(3,5,7-trihydroxy-4-oxo-4H-chromen-2-yl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (1)
3.3. Pharmacological Analysis
3.3.1. Animals
3.3.2. Cell Isolation Procedure
3.3.3. Whole-Cell Patch Clamp Recordings
3.3.4. IKCa1.1 Current Measurement
3.3.5. IBa1.2 Recording
3.3.6. Drugs and Chemicals
3.3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patel, R.V.; Mistry, B.M.; Shinde, S.K.; Syed, R.; Singh, V.; Shin, H.-S. Therapeutic potential of quercetin as a cardiovascular agent. Eur. J. Med. Chem. 2018, 155, 889–904. [Google Scholar] [CrossRef]
- Scholz, E.P.; Zitron, E.; Katus, H.A.; Karle, C.A. Cardiovascular ion channels as a molecular target of flavonoids. Cardiovasc. Ther. 2010, 28, 46–52. [Google Scholar] [CrossRef]
- Saponara, S.; Carosati, E.; Mugnai, P.; Sgaragli, G.; Fusi, F. The flavonoid scaffold as a template for the design of modulators of the vascular Ca(v) 1.2 channels. Br. J. Pharmacol. 2011, 164, 1684–1697. [Google Scholar] [CrossRef] [Green Version]
- Fusi, F.; Spiga, O.; Trezza, A.; Sgaragli, G.; Saponara, S. The surge of flavonoids as novel, fine regulators of cardiovascular Cav channels. Eur. J. Pharmacol. 2017, 796, 158–174. [Google Scholar] [CrossRef]
- Fusi, F.; Trezza, A.; Tramaglino, M.; Sgaragli, G.; Saponara, S.; Spiga, O. The beneficial health effects of flavonoids on the cardiovascular system: Focus on K+ channels. Pharmacol. Res. 2020, 152, 104625. [Google Scholar] [CrossRef]
- Saponara, S.; Sgaragli, G.; Fusi, F. Quercetin as a novel activator of L-type Ca(2+) channels in rat tail artery smooth muscle cells. Br. J. Pharmacol. 2002, 135, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Iozzi, D.; Schubert, R.; Kalenchuk, V.U.; Neri, A.; Sgaragli, G.; Fusi, F.; Saponara, S. Quercetin relaxes rat tail main artery partly via a PKG-mediated stimulation of KCa 1.1 channels. Acta Physiol. (Oxf.) 2013, 208, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.; Palade, P.T.; Marsh, J.D.; Telemaque, S.; Pesic, A.; Rusch, N.J. Vascular calcium channels and high blood pressure: Pathophysiology and therapeutic implications. Vasc. Pharmacol. 2006, 44, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.H.; Rusch, N.J. New expression profiles of voltage-gated ion channels in arteries exposed to high blood pressure. Microcirculation 2002, 9, 243–257. [Google Scholar] [CrossRef]
- Magar, R.T.; Sohng, J.K. A review on structure, modifications and structure-activity relation of quercetin and its derivatives. J. Microbiol. Biotechnol. 2020, 30, 11–20. [Google Scholar] [CrossRef]
- Badolato, M.; Carullo, G.; Perri, M.; Cione, E.; Manetti, F.; Di Gioia, M.L.; Brizzi, A.; Caroleo, M.C.; Aiello, F. Quercetin/oleic acid-based G-protein-coupled receptor 40 ligands as new insulin secretion modulators. Future Med. Chem. 2017, 9, 1873–1885. [Google Scholar] [CrossRef]
- Carullo, G.; Ahmed, A.; Trezza, A.; Spiga, O.; Brizzi, A.; Saponara, S.; Fusi, F.; Aiello, F. Design, synthesis and pharmacological evaluation of ester-based quercetin derivatives as selective vascular KCa1.1 channel stimulators. Bioorg. Chem. 2020, 105, 104404. [Google Scholar] [CrossRef]
- Carullo, G.; Ahmed, A.; Trezza, A.; Spiga, O.; Brizzi, A.; Saponara, S.; Fusi, F.; Aiello, F. A multitarget semi-synthetic derivative of the flavonoid morin with improved in vitro vasorelaxant activity: Role of CaV1.2 and KCa1.1 channels. Biochem. Pharmacol. 2021, 185, 114429. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, S.; Governa, P.; Borgonetti, V.; Marcolongo, P.; Nanni, C.; Gamberucci, A.; Manetti, F.; Pessina, F.; Carullo, G.; Brizzi, A.; et al. Pinocembrin and its linolenoyl ester derivative induce wound healing activity in HaCaT cell line potentially involving a GPR120/FFA4 mediated pathway. Bioorg. Chem. 2021, 108, 104657. [Google Scholar] [CrossRef]
- Chaudhary, A.; Jaswal, V.S.; Choudhary, S.; Sonika; Sharma, A.; Beniwal, V.; Tuli, H.S.; Sharma, S. Ferulic acid: A promising therapeutic phytochemical and recent patents advances. Recent Pat. Inflamm. Allergy Drug Discov. 2019, 13, 115–123. [Google Scholar] [CrossRef]
- Fuentes, E.; Palomo, I. Mechanisms of endothelial cell protection by hydroxycinnamic acids. Vasc. Pharmacol. 2014, 63, 155–161. [Google Scholar] [CrossRef]
- Niu, X.; Qian, X.; Magleby, K.L. Linker-gating ring complex as passive spring and Ca(2+)-dependent machine for a voltage- and Ca(2+)-activated potassium channel. Neuron 2004, 42, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Gessner, G.; Cui, Y.M.; Otani, Y.; Ohwada, T.; Soom, M.; Hoshi, T.; Heinemann, S.H. Molecular mechanism of pharmacological activation of BK channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3552–3557. [Google Scholar] [CrossRef] [Green Version]
- Bouktaib, M.; Lebrun, S.; Atmani, A.; Rolando, C. Hemisynthesis of all the O-monomethylated analogues of quercetin including the major metabolites, through selective protection of phenolic functions. Tetrahedron 2002, 58, 10001–10009. [Google Scholar] [CrossRef]
- Trouillas, P.; Marsal, P.; Siri, D.; Lazzaroni, R.; Duroux, J.-L. A DFT study of the reactivity of OH groups in quercetin and taxifolin antioxidants: The specificity of the 3-OH site. Food Chem. 2006, 97, 679–688. [Google Scholar] [CrossRef]
- Veverka, M.; Gallovič, J.; Švajdlenka, E.; Ververková, E.; Prónayová, N.; Miláčková, I.; Štefek, M. Novel quercetin derivatives: Synthesis and screening for anti-oxidant activity and aldose reductase inhibition. Chem. Pap. 2013, 67, 76–83. [Google Scholar] [CrossRef]
- Salem, J.H.; Humeau, C.; Chevalot, I.; Harscoat-Schiavo, C.; Vanderesse, R.; Blanchard, F.; Fick, M. Effect of acyl donor chain length on isoquercitrin acylation and biological activities of corresponding esters. Process Biochem. 2010, 45, 382–389. [Google Scholar] [CrossRef]
- Saik, A.Y.H.; Lim, Y.Y.; Stanslas, J.; Choo, W.M. Enzymatic synthesis of quercetin oleate esters using Candida antarctica lipase B. Biotechnol. Lett. 2017, 39, 297–304. [Google Scholar] [CrossRef]
- Soltesova-Prnova, M.; Milackova, I.; Stefek, M. 3’-O-(3-Chloropivaloyl)quercetin, α-glucosidase inhibitor with multi-targeted therapeutic potential in relation to diabetic complications. Chem. Pap. 2016, 70, 1439–1444. [Google Scholar] [CrossRef]
- Kumar, V.; Jahan, F.; Mahajan, R.V.; Kumar Saxena, R. Efficient regioselective acylation of quercetin using Rhizopus oryzae lipase and its potential as antioxidant. Bioresour. Technol. 2016, 218, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Russo, N.; Toscano, M.; Uccella, N. Semiempirical Molecular Modeling into Quercetin Reactive Site: Structural, Conformational, and Electronic Features. J. Agric. Food Chem. 2000, 48, 3232–3237. [Google Scholar] [CrossRef]
- Chebil, L.; Anthoni, J.; Humeau, C.; Gerardin, C.; Engasser, J.-M.; Ghoul, M. Enzymatic Acylation of Flavonoids: Effect of the Nature of the Substrate, Origin of Lipase, and Operating Conditions on Conversion Yield and Regioselectivity. J. Agric. Food Chem. 2007, 55, 9496–9502. [Google Scholar] [CrossRef] [PubMed]
- Saik, A.Y.H.; Lim, Y.Y.; Stanslas, J.; Choo, W.S. Lipase-catalyzed acylation of quercetin with cinnamic acid. Biocatal. Biotransform. 2016, 34, 33–43. [Google Scholar] [CrossRef]
- Kyriakou, E.; Primikyri, A.; Charisiadis, P.; Katsoura, M.; Gerothanassis, I.P.; Stamatis, H.; Tzakos, A.G. Unexpected enzyme-catalyzed regioselective acylation of flavonoid aglycones and rapid product screening. Org. Biomol. Chem. 2012, 10, 1739. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Fusi, F.; Saponara, S.; Sgaragli, G.; Cargnelli, G.; Bova, S. Ca(2+) entry blocking and contractility promoting actions of norbormide in single rat caudal artery myocytes. Br. J. Pharmacol. 2002, 137, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Mugnai, P.; Durante, M.; Sgaragli, G.; Saponara, S.; Paliuri, G.; Bova, S.; Fusi, F. L-type Ca(2+) channel current characteristics are preserved in rat tail artery myocytes after one-day storage. Acta Physiol. (Oxf.) 2014, 211, 334–345. [Google Scholar] [CrossRef]
- Fusi, F.; Sgaragli, G.; Saponara, S. Mechanism of myricetin stimulation of vascular L-type Ca2+ current. J. Pharmacol. Exp. Ther. 2005, 313, 790–797. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogen/Carbon Position | Que δ | Compd. 1 δ |
|---|---|---|
| C in 3 1 | 136.2 | 136.6 |
| C in 5 | 161.2 | 161.2 |
| C-H in 6 | 6.18/98.7 | 6.19/98.7 |
| C in 7 | 164.3 | 164.5 |
| C-H in 8 | 6.40/93.8 | 6.46/94.1 |
| C-H in 2′ | 7.67/115.5 | 7.92/123.3 |
| C in 3′ | 145.5 | 138.8 |
| C in 4′ | 148.2 | 151.6 |
| C-H in 5′ | 6.88/116.1 | 7.11/117.3 |
| C-H in 6′ | 7.53/120.4 | 7.96/126.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brizzi, A.; Trezza, A.; Spiga, O.; Maramai, S.; Scorzelli, F.; Saponara, S.; Fusi, F. 2-Hydroxy-5-(3,5,7-trihydroxy-4-oxo-4H-chromen-2-yl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate: Synthesis, In Silico Analysis and In Vitro Pharmacological Evaluation. Molbank 2021, 2021, M1258. https://doi.org/10.3390/M1258
Brizzi A, Trezza A, Spiga O, Maramai S, Scorzelli F, Saponara S, Fusi F. 2-Hydroxy-5-(3,5,7-trihydroxy-4-oxo-4H-chromen-2-yl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate: Synthesis, In Silico Analysis and In Vitro Pharmacological Evaluation. Molbank. 2021; 2021(3):M1258. https://doi.org/10.3390/M1258
Chicago/Turabian StyleBrizzi, Antonella, Alfonso Trezza, Ottavia Spiga, Samuele Maramai, Francesco Scorzelli, Simona Saponara, and Fabio Fusi. 2021. "2-Hydroxy-5-(3,5,7-trihydroxy-4-oxo-4H-chromen-2-yl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate: Synthesis, In Silico Analysis and In Vitro Pharmacological Evaluation" Molbank 2021, no. 3: M1258. https://doi.org/10.3390/M1258
APA StyleBrizzi, A., Trezza, A., Spiga, O., Maramai, S., Scorzelli, F., Saponara, S., & Fusi, F. (2021). 2-Hydroxy-5-(3,5,7-trihydroxy-4-oxo-4H-chromen-2-yl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate: Synthesis, In Silico Analysis and In Vitro Pharmacological Evaluation. Molbank, 2021(3), M1258. https://doi.org/10.3390/M1258