
Cyclo-Tetrakis(μ-diphenylphosphido)-1,5-bis(tri-tert-butylphosphine)-Tetracopper



Abstract
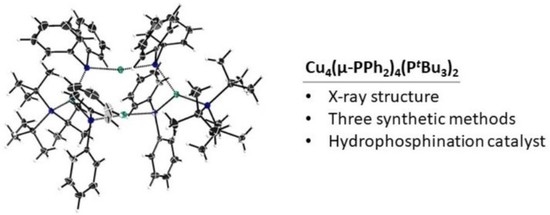
:
1. Introduction
2. Results and Discussion


3. Experimental Details
3.1. General Considerations
3.2. Synthesis of Compound 1
3.3. X-ray Structure Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waterman, R. Metal-phosphido and -phosphinidene complexes in P–E bond-forming reactions. Dalton Trans. 2009, 1, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Glueck, D.S. Metal-Catalyzed P–C Bond Formation via P–H Oxidative Addition: Fundamentals and Recent Advances. J. Org. Chem. 2020, 85, 14276–14285. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L. Mechanisms of Metal-Catalyzed Hydrophosphination of Alkenes and Alkynes. ACS Catal. 2013, 3, 2845–2855. [Google Scholar] [CrossRef]
- Cowley, A.H.; Giolando, D.M.; Jones, R.A.; Nunn, C.M.; Power, J.M. A tetrameric copper(I) phosphide and its conversion into a bis(phosphido)cuprate and a bis(phosphine)-complexed tetrameric copper(I) thiolate. J. Chem. Soc. Chem. Commun. 1988, 3, 208–209. [Google Scholar] [CrossRef]
- Eichhöfer, A.; Fenske, D.; Holstein, W. New Phosphido-Bridging Copper Clusters. Angew. Chem. Int. Ed. 1993, 32, 242–245. [Google Scholar] [CrossRef]
- Fenske, D.; Holstein, W. [Cu96P30{P(SiMe3)2}6(PEt3)18], a New Phosphorus-Bridged Copper Cluster. Angew. Chem. Int. Ed. 1994, 33, 1290–1292. [Google Scholar] [CrossRef]
- Annan, T.A.; Kumar, R.; Tuck, D.G. The direct electrochemical synthesis of metal–diphenylphosphido complexes, and the crystal structure of Cu4(µ-PPh2)4(Ph2PCH2PPh2)2. J. Chem. Soc. Chem. Commun. 1988, 6, 446–448. [Google Scholar] [CrossRef]
- Annan, T.A.; Kumar, R.; Tuck, D.G. Direct electrochemical synthesis and crystallographic characterization of metal diphenylphosphido and diphenylthiophosphinato compounds, and some derivatives. J. Chem. Soc. Dalton Trans. 1991, 1, 11–18. [Google Scholar] [CrossRef]
- Meyer, C.; Grützmacher, H.; Pritzkow, H. Copper Pnictogenides as Selective Reagents: A New Access to Functionalized Phosphanes and Arsanes. Angew. Chem. Int. Ed. 1997, 36, 2471–2473. [Google Scholar] [CrossRef]
- Brauer, D.J.; Hessler, G.; Knueppel, P.C.; Stelzer, O. Linear oligophosphaalkanes. 23. Novel copper(I) complexes with electron-deficient triply bridging secondary phosphido groups.mu.3-PRR′ (R = isopropyl, tert-butyl, R′ = CH2-P R2; R = R′ = phenyl). Inorg. Chem. 1990, 29, 2370–2375. [Google Scholar] [CrossRef]
- Gómez-Ruiz, S.; Wolf, R.; Bauer, S.; Bittig, H.; Schisler, A.; Lönnecke, P.; Hey-Hawkins, E. Coordination Chemistry of the cyclo-(P5tBu4)− Ion: Monomeric and Oligomeric Copper(I), Silver(I) and Gold(I) Complexes. Chem. Eur. J. 2008, 14, 4511–4520. [Google Scholar] [CrossRef]
- Gómez-Ruiz, S.; Wolf, R.; Hey-Hawkins, E. Different transmetallation behaviour of [M(P4HR4)] salts toward rhodium(i) and copper(i) (M = Na, K; R = Ph, Mes; Mes = 2,4,6-Me3C6H2). Dalton Trans. 2008, 15, 1982–1988. [Google Scholar] [CrossRef]
- Ziółkowska, A.; Brauer, S.; Ponikiewski, Ł. First Copper(I) and Silver(I) Complexes Containing Phosphanylphosphido Ligands. Z. Anorg. Allg. Chem. 2019, 645, 949–954. [Google Scholar] [CrossRef]
- Scholz, S.; Bolte, M.; Wagner, M.; Lerner, H.-W. Synthese und Eigenschaften der Di-tert-butylphosphide [M(PtBu2)2]2 (M = Zn, Hg) und [Cu(PtBu2)]4. Z. Anorg. Allg. Chem. 2007, 633, 1199–1204. [Google Scholar] [CrossRef]
- Yuan, J.; Zhu, L.; Zhang, J.; Li, J.; Cui, C. Sequential Addition of Phosphine to Alkynes for the Selective Synthesis of 1,2-Diphosphinoethanes under Catalysis. Well-Defined NHC-Copper Phosphides vs in Situ CuCl2/NHC Catalyst. Organometallics 2017, 36, 455–459. [Google Scholar] [CrossRef]
- Khalili Najafabadi, B.; Corrigan, J.F. Enhanced thermal stability of Cu–silylphosphido complexes via NHC ligation. Dalton Trans. 2015, 44, 14235–14241. [Google Scholar] [CrossRef]
- Horsley Downie, T.M.; Hall, J.W.; Collier Finn, T.P.; Liptrot, D.J.; Lowe, J.P.; Mahon, M.F.; McMullin, C.L.; Whittlesey, M.K. The first ring-expanded NHC–copper(i) phosphides as catalysts in the highly selective hydrophosphination of isocyanates. ChemComm 2020, 56, 13359–13362. [Google Scholar] [CrossRef]
- Dannenberg, S.G.; Waterman, R. A bench-stable copper photocatalyst for the rapid hydrophosphination of activated and unactivated alkenes. ChemComm 2020, 56, 14219–14222. [Google Scholar] [CrossRef]
- Huggins, M.T.; Kesharwani, T.; Buttrick, J.; Nicholson, C. Variable Temperature NMR Experiment Studying Restricted Bond Rotation. J. Chem. Ed. 2020, 97, 1425–1429. [Google Scholar] [CrossRef]
- Bianco, V.D.; Doronzo, S.; Chan, J.; Bennett, M.A. Diphenylphosphine. In Inorganic Syntheses; McGraw-Hill, Inc.: New York, NY, USA, 1976; pp. 161–163. [Google Scholar]
- Keller, R.N.; Wrcoff, H.D.; Marchi, L.E. Copper(I) Chloride. In Inorganic Syntheses; McGraw-Hill, Inc.: New York, NY, USA, 1946; pp. 1–4. [Google Scholar]
- Tsuda, T.; Yazawa, T.; Watanabe, K.; Fujii, T.; Saegusa, T. Preparation of thermally stable and soluble mesitylcopper(I) and its application in organic synthesis. J. Org. Chem. 1981, 46, 192–194. [Google Scholar] [CrossRef]
- Hanawalt, E.M.; Farkas, J.; Richey, H.G. Organomagnesates from Reactions of Dialkylmagnesium Compounds with Alkali-Metal Alkoxides, Potassium Hydride, and Other Salts. Organometallics 2004, 23, 416–422. [Google Scholar] [CrossRef]
- Novas, B.T.; Bange, C.A.; Waterman, R. Photocatalytic Hydrophosphination of Alkenes and Alkynes Using Diphenylphosphine and Triamidoamine-Supported Zirconium. Eur. J. Inorg. Chem. 2019, 2019, 1640–1643. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Atom–Atom | Length [Å] | Atom–Atom–Atom | Angle [°] |
|---|---|---|---|
| Cu1–P5 | 2.2738(10) | P5–Cu1–P1 | 130.14(4) |
| Cu1–P1 | 2.3076(10) | P4–Cu1–P1 | 98.75(4) |
| P1–Cu2 | 2.2272(12) | Cu2–P1–Cu1 | 115.59(4) |
| Cu2–Cu4 | 2.8612(13) | P2–Cu2–P1 | 167.32(3) |
| P2–Cu2–Cu4 | 96.16(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dannenberg, S.G.; Waterman, R. Cyclo-Tetrakis(μ-diphenylphosphido)-1,5-bis(tri-tert-butylphosphine)-Tetracopper. Molbank 2022, 2022, M1334. https://doi.org/10.3390/M1334
Dannenberg SG, Waterman R. Cyclo-Tetrakis(μ-diphenylphosphido)-1,5-bis(tri-tert-butylphosphine)-Tetracopper. Molbank. 2022; 2022(1):M1334. https://doi.org/10.3390/M1334
Chicago/Turabian StyleDannenberg, Steven G., and Rory Waterman. 2022. "Cyclo-Tetrakis(μ-diphenylphosphido)-1,5-bis(tri-tert-butylphosphine)-Tetracopper" Molbank 2022, no. 1: M1334. https://doi.org/10.3390/M1334
APA StyleDannenberg, S. G., & Waterman, R. (2022). Cyclo-Tetrakis(μ-diphenylphosphido)-1,5-bis(tri-tert-butylphosphine)-Tetracopper. Molbank, 2022(1), M1334. https://doi.org/10.3390/M1334