The Inflammasome in Host Defense

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Inflammasome
2.1. The NLRP1/NALP1 Inflammasome
2.2. The NLRC4/Ipaf Inflammasome
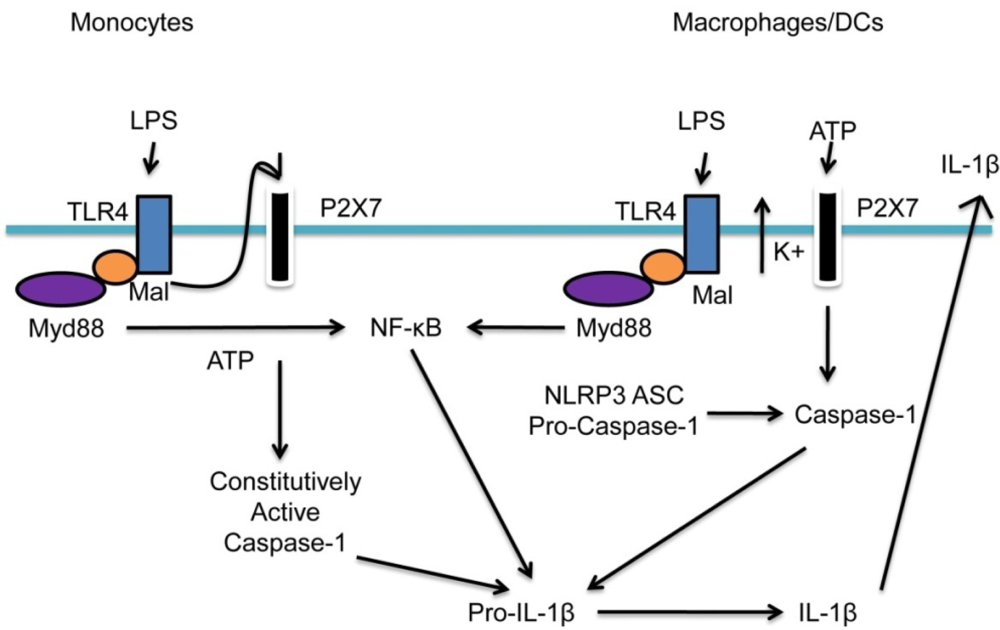
2.3. The NLRP3/NALP3 Inflammasome
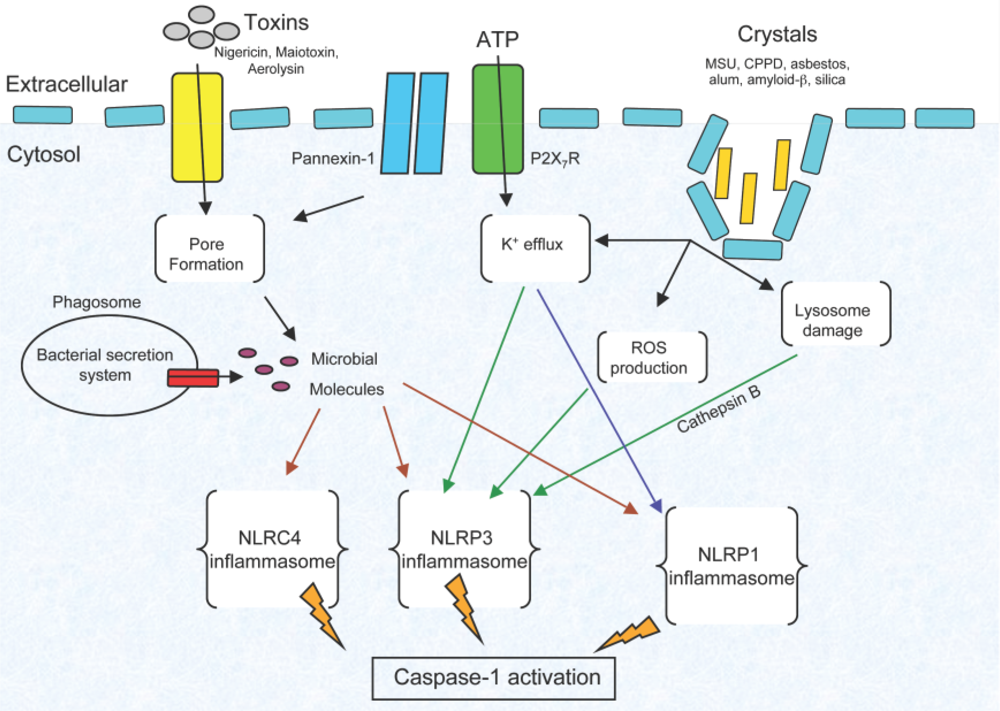
3. Inflammasome Activation Requirements
4. Conclusions
Acknowledgments
Abbreviations
| TLRs | Toll-like receptors |
| RLRs | Retinoic acid-inducible gene I-like receptors |
| NLRs | Nod-like receptors |
| IL | interleukin |
| CARD | Caspase recruitment domain |
| ASC | apoptosis associated speck-like protein containing a CARD |
| TIR | Toll-interleukin-1 receptor domain |
| BIR | baculovirus IAP (inhibitor of apoptosis protein) repeat |
| NLRC4/IPAF | interleukin 1β-converting enzyme protease-activating factor |
| LRR | leucine-rich repeat |
| NACHT | domain present in NAIP, CIITA, HET-E (Podospora anserine incompatibility locus protein) and telomerase associated protein |
| NAD | NACHT-associated domain; NAIP–neuronal apoptosis inhibitor protein |
| PYD | pyrin domain |
| GBP | GTP binding domain |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| ESX-1 | (ESAT-6 secretion system-1) |
| ROS | Reactive oxygen species |
| LPS | lipopolysaccharide. |
References and Notes
- Palm, N.W.; Medzhitov, R. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev 2009, 227, 221–233. [Google Scholar]
- Medzhitov, R. Approaching the asymptote: 20 years later. Immunity 2009, 30, 766–775. [Google Scholar]
- Creagh, E.M.; O’Neill, L.A. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol 2006, 27, 352–357. [Google Scholar]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar]
- van Duin, D.; Medzhitov, R.; Shaw, A.C. Triggering TLR signaling in vaccination. Trends Immunol 2006, 27, 49–55. [Google Scholar]
- Nakhaei, P.; Genin, P.; Civas, A.; Hiscott, J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin. Immunol 2009, 21, 215–222. [Google Scholar]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci 2008, 1143, 1–20. [Google Scholar]
- Tiemi Shio, M.; Eisenbarth, S.C.; Savaria, M.; Vinet, A.F.; Bellemare, M.J.; Harder, K.W.; Sutterwala, F.S.; Bohle, D.S.; Descoteaux, A.; Flavell, R.A.; Olivier, M. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog 2009, 5, e1000559. [Google Scholar]
- Brodsky, I.E.; Monack, D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin. Immunol 2009, 21, 199–207. [Google Scholar]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol 2009, 27, 229–265. [Google Scholar]
- Pedra, J.H.; Cassel, S.L.; Sutterwala, F.S. Sensing pathogens and danger signals by the inflammasome. Cur. Opin. Immunol 2009, 21, 10–16. [Google Scholar]
- Shirasu, K. The HSP90-SGT1 chaperone complex for NLR immune sensors. Annu. Rev. Plant Biol 2009, 60, 139–164. [Google Scholar]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; Hoffman, H.M.; Hugot, J.P.; Inohara, N.; Mackenzie, A.; Maltais, L.J.; Nunez, G.; Ogura, Y.; Otten, L.A.; Philpott, D.; Reed, J.C.; Reith, W.; Schreiber, S.; Steimle, V.; Ward, P.A. The NLR gene family: a standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar]
- Proell, M.; Riedl, S.J.; Fritz, J.H.; Rojas, A.M.; Schwarzenbacher, R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One 2008, 3, e2119. [Google Scholar]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mole. Cell 2007, 25, 713–724. [Google Scholar]
- Wickliffe, K.E.; Leppla, S.H.; Moayeri, M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell. Microbiol 2008, 10, 332–343. [Google Scholar]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet 2006, 38, 240–244. [Google Scholar]
- Reig, N.; Jiang, A.; Couture, R.; Sutterwala, F.S.; Ogura, Y.; Flavell, R.A.; Mellman, I.; van der Goot, F.G. Maturation modulates caspase-1-independent responses of dendritic cells to Anthrax lethal toxin. Cell. Microbiol 2008, 10, 1190–1207. [Google Scholar]
- Squires, R.C.; Muehlbauer, S.M.; Brojatsch, J. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J. Biol. Chem 2007, 282, 34260–34267. [Google Scholar]
- Liao, K.C.; Mogridge, J. Expression of Nlrp1b inflammasome components in human fibroblasts confers susceptibility to anthrax lethal toxin. Infect. Immun 2009, 77, 4455–4462. [Google Scholar]
- Newman, Z.L.; Leppla, S.H.; Moayeri, M. CA-074Me protection against anthrax lethal toxin. Infect. Immun 2009, 77, 4327–4336. [Google Scholar]
- Nour, A.M.; Yeung, Y.G.; Santambrogio, L.; Boyden, E.D.; Stanley, E.R.; Brojatsch, J. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect. Immun 2009, 77, 1262–1271. [Google Scholar]
- Hsu, L.C.; Ali, S.R.; McGillivray, S.; Tseng, P.H.; Mariathasan, S.; Humke, E.W.; Eckmann, L.; Powell, J.J.; Nizet, V.; Dixit, V.M.; Karin, M. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA 2008, 105, 7803–7808. [Google Scholar]
- Guarda, G.; Dostert, C.; Staehli, F.; Cabalzar, K.; Castillo, R.; Tardivel, A.; Schneider, P.; Tschopp, J. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 2009, 460, 269–273. [Google Scholar]
- Amer, A.; Franchi, L.; Kanneganti, T.D.; Body-Malapel, M.; Ozoren, N.; Brady, G.; Meshinchi, S.; Jagirdar, R.; Gewirtz, A.; Akira, S.; Nunez, G. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem 2006, 281, 35217–35223. [Google Scholar]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; Grant, E.P.; Nunez, G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat. Immunol 2006, 7, 576–582. [Google Scholar]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar]
- Franchi, L.; Stoolman, J.; Kanneganti, T.D.; Verma, A.; Ramphal, R.; Nunez, G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur. J. Immunol 2007, 37, 3030–3039. [Google Scholar]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol 2006, 7, 569–575. [Google Scholar]
- Warren, S.E.; Mao, D.P.; Rodriguez, A.E.; Miao, E.A.; Aderem, A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol 2008, 180, 7558–7564. [Google Scholar]
- Fink, S.L.; Bergsbaken, T.; Cookson, B.T. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 4312–4317. [Google Scholar]
- Miao, E.A.; Ernst, R.K.; Dors, M.; Mao, D.P.; Aderem, A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. USA 2008, 105, 2562–2567. [Google Scholar]
- Sutterwala, F.S.; Mijares, L.A.; Li, L.; Ogura, Y.; Kazmierczak, B.I.; Flavell, R.A. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med 2007, 204, 3235–3245. [Google Scholar]
- Franchi, L.; Stoolman, J.; Kanneganti, T.D.; Verma, A.; Ramphal, R.; Nunez, G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur. J. Immunol 2007, 37, 3030–3039. [Google Scholar]
- Suzuki, T.; Nunez, G. A role for Nod-like receptors in autophagy induced by Shigella infection. Autophagy 2008, 4, 73–75. [Google Scholar]
- Suzuki, T.; Franchi, L.; Toma, C.; Ashida, H.; Ogawa, M.; Yoshikawa, Y.; Mimuro, H.; Inohara, N.; Sasakawa, C.; Nunez, G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 2007, 3, e111. [Google Scholar]
- Pedra, J.H.; Sutterwala, F.S.; Sukumaran, B.; Ogura, Y.; Qian, F.; Montgomery, R.R.; Flavell, R.A.; Fikrig, E. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-{gamma} axis during anaplasma phagocytophilum infection. J. Immunol 2007, 179, 4783–4791. [Google Scholar]
- Sun, Y.H.; Rolan, H.G.; Tsolis, R.M. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype typhimurium. J. Biol. Chem 2007, 282, 33897–33901. [Google Scholar]
- Diez, E.; Lee, S.H.; Gauthier, S.; Yaraghi, Z.; Tremblay, M.; Vidal, S.; Gros, P. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat. Genet 2003, 33, 55–60. [Google Scholar]
- Growney, J.D.; Dietrich, W.F. High-resolution genetic and physical map of the Lgn1 interval in C57BL/6J implicates Naip2 or Naip5 in Legionella pneumophila pathogenesis. Genome Res 2000, 10, 1158–1171. [Google Scholar]
- Wright, E.K.; Goodart, S.A.; Growney, J.D.; Hadinoto, V.; Endrizzi, M.G.; Long, E.M.; Sadigh, K.; Abney, A.L.; Bernstein-Hanley, I.; Dietrich, W.F. Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol 2003, 13, 27–36. [Google Scholar]
- Zamboni, D.S.; Kobayashi, K.S.; Kohlsdorf, T.; Ogura, Y.; Long, E.M.; Vance, R.E.; Kuida, K.; Mariathasan, S.; Dixit, V.M.; Flavell, R.A.; Dietrich, W.F.; Roy, C.R. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol 2006, 7, 318–325. [Google Scholar]
- Ren, T.; Zamboni, D.S.; Roy, C.R.; Dietrich, W.F.; Vance, R.E. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog 2006, 2, e18. [Google Scholar]
- Molofsky, A.B.; Byrne, B.G.; Whitfield, N.N.; Madigan, C.A.; Fuse, E.T.; Tateda, K.; Swanson, M.S. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exper. Med 2006, 203, 1093–1104. [Google Scholar]
- Lamkanfi, M.; Amer, A.; Kanneganti, T.D.; Munoz-Planillo, R.; Chen, G.; Vandenabeele, P.; Fortier, A.; Gros, P.; Nunez, G. The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol 2007, 178, 8022–8027. [Google Scholar]
- Lightfield, K.L.; Persson, J.; Brubaker, S.W.; Witte, C.E.; von Moltke, J.; Dunipace, E.A.; Henry, T.; Sun, Y.H.; Cado, D.; Dietrich, W.F.; Monack, D.M.; Tsolis, R.M.; Vance, R.E. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol 2008, 9, 1171–1178. [Google Scholar]
- Akhter, A.; Gavrilin, M.A.; Frantz, L.; Washington, S.; Ditty, C.; Limoli, D.; Day, C.; Sarkar, A.; Newland, C.; Butchar, J.; Marsh, C.B.; Wewers, M.D.; Tridandapani, S.; Kanneganti, T.D.; Amer, A.O. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog 2009, 5, e1000361. [Google Scholar]
- Lamkanfi, M.; Malireddi, R.K.; Kanneganti, T.D. Fungal zymosan and mannan activate the cryopyrin inflammasome. J. Biol. Chem 2009, 284, 20574–20581. [Google Scholar]
- Lamkanfi, M.; Kanneganti, T.D.; Van Damme, P.; Vanden Berghe, T.; Vanoverberghe, I.; Vandekerckhove, J.; Vandenabeele, P.; Gevaert, K.; Nunez, G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteomics 2008, 7, 2350–2363. [Google Scholar]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol 2008, 9, 866–872. [Google Scholar]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar]
- Sutterwala, F.S.; Ogura, Y.; Szczepanik, M.; Lara-Tejero, M.; Lichtenberger, G.S.; Grant, E.P.; Bertin, J.; Coyle, A.J.; Galan, J.E.; Askenase, P.W.; Flavell, R.A. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006, 24, 317–327. [Google Scholar]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar]
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; Bertin, J.; Coyle, A.; Grant, E.P.; Akira, S.; Nunez, G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar]
- Willingham, S.B.; Bergstralh, D.T.; O’Connor, W.; Morrison, A.C.; Taxman, D.J.; Duncan, J.A.; Barnoy, S.; Venkatesan, M.M.; Flavell, R.A.; Deshmukh, M.; Hoffman, H.M.; Ting, J.P. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2007, 2, 147–159. [Google Scholar]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol 2008. [Google Scholar]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol 2008. [Google Scholar]
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.L.; Stehle, J.C.; Kopf, M.; Stamenkovic, I.; Corradin, G.; Tschopp, J. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 2009, 4, e6510. [Google Scholar]
- Bostanci, N.; Emingil, G.; Saygan, B.; Turkoglu, O.; Atilla, G.; Curtis, M.A.; Belibasakis, G.N. Expression and regulation of the NALP3 inflammasome complex in periodontal diseases. Clin. Exp. Immunol 2009, 157, 415–422. [Google Scholar]
- Huang, M.T.; Taxman, D.J.; Holley-Guthrie, E.A.; Moore, C.B.; Willingham, S.B.; Madden, V.; Parsons, R.K.; Featherstone, G.L.; Arnold, R.R.; O’Connor, B.P.; Ting, J.P. Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by porphyromonas gingivalis in human cells. J. Immunol 2009, 182, 2395–2404. [Google Scholar]
- Duncan, J.A.; Gao, X.; Huang, M.T.; O’Connor, B.P.; Thomas, C.E.; Willingham, S.B.; Bergstralh, D.T.; Jarvis, G.A.; Sparling, P.F.; Ting, J.P. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol 2009, 182, 6460–6469. [Google Scholar]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; Doherty, P.C.; Kanneganti, T.D. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar]
- Abdul-Sater, A.A.; Koo, E.; Hacker, G.; Ojcius, D.M. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen chlamydia trachomatis. J. Biol. Chem 2009. [Google Scholar]
- Kanneganti, T.D.; Lamkanfi, M.; Kim, Y.G.; Chen, G.; Park, J.H.; Franchi, L.; Vandenabeele, P.; Nunez, G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 2007, 26, 433–443. [Google Scholar]
- Pelegrin, P.; Barroso-Gutierrez, C.; Surprenant, A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J. Immunol 2008, 180, 7147–7157. [Google Scholar]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J 2006, 25, 5071–5082. [Google Scholar]
- Pelegrin, P.; Surprenant, A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J. Biol. Chem 2007, 282, 2386–2394. [Google Scholar]
- Koo, I.C.; Wang, C.; Raghavan, S.; Morisaki, J.H.; Cox, J.S.; Brown, E.J. ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell. Microbiol 2008. [Google Scholar]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; Superti-Furga, G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol 2009, 10, 266–272. [Google Scholar]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; Hume, D.A.; Stacey, K.J. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar]
- Kankkunen, P.; Rintahaka, J.; Aalto, A.; Leino, M.; Majuri, M.L.; Alenius, H.; Wolff, H.; Matikainen, S. Trichothecene mycotoxins activate inflammatory response in human macrophages. J. Immunol 2009, 182, 6418–6425. [Google Scholar]
- Gross, O.; Poeck, H.; Bscheider, M.; Dostert, C.; Hannesschlager, N.; Endres, S.; Hartmann, G.; Tardivel, A.; Schweighoffer, E.; Tybulewicz, V.; Mocsai, A.; Tschopp, J.; Ruland, J. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009, 459, 433–436. [Google Scholar]
- Joly, S.; Ma, N.; Sadler, J.J.; Soll, D.R.; Cassel, S.L.; Sutterwala, F.S. Cutting Edge: Candida albicans Hyphae Formation Triggers Activation of the Nlrp3 Inflammasome. J. Immunol 2009. [Google Scholar]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar]
- van de Veerdonk, F.L.; Joosten, L.A.; Devesa, I.; Mora-Montes, H.M.; Kanneganti, T.D.; Dinarello, C.A.; van der Meer, J.W.; Gow, N.A.; Kullberg, B.J.; Netea, M.G. Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J. Infect. Dis 2009, 199, 1087–1096. [Google Scholar]
- Saito, M.; Nishikomori, R.; Kambe, N.; Fujisawa, A.; Tanizaki, H.; Takeichi, K.; Imagawa, T.; Iehara, T.; Takada, H.; Matsubayashi, T.; Tanaka, H.; Kawashima, H.; Kawakami, K.; Kagami, S.; Okafuji, I.; Yoshioka, T.; Adachi, S.; Heike, T.; Miyachi, Y.; Nakahata, T. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood 2008, 111, 2132–2141. [Google Scholar]
- Fujisawa, A.; Kambe, N.; Saito, M.; Nishikomori, R.; Tanizaki, H.; Kanazawa, N.; Adachi, S.; Heike, T.; Sagara, J.; Suda, T.; Nakahata, T.; Miyachi, Y. Disease-associated mutations in CIAS1 induce cathepsin B-dependent rapid cell death of human THP-1 monocytic cells. Blood 2007, 109, 2903–2911. [Google Scholar]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007, 14, 1583–1589. [Google Scholar]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem 2007, 282, 2871–2879. [Google Scholar]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar]
- Ng, G.; Sharma, K.; Ward, S.M.; Desrosiers, M.D.; Stephens, L.A.; Schoel, W.M.; Li, T.; Lowell, C.A.; Ling, C.C.; Amrein, M.W.; Shi, Y. Receptor-independent, direct membrane binding leads to cell-surface lipid sorting and syk kinase activation in dendritic cells. Immunity 2008, 29, 807–818. [Google Scholar]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglie, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad Sci USA 2008, 105, 8067–8072. [Google Scholar]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; Funk, C.J.; Mason, R.J.; Kullberg, B.J.; Rubartelli, A.; van der Meer, J.W.; Dinarello, C.A. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem 2009, 284, 18143–18151. [Google Scholar]
- Nakamura, Y.; Kambe, N.; Saito, M.; Nishikomori, R.; Kim, Y.G.; Murakami, M.; Nunez, G.; Matsue, H. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J. Exp. Med 2009, 206, 1037–1046. [Google Scholar]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; Perfettini, J.L.; Schlemmer, F.; Tasdemir, E.; Uhl, M.; Genin, P.; Civas, A.; Ryffel, B.; Kanellopoulos, J.; Tschopp, J.; Andre, F.; Lidereau, R.; McLaughlin, N.M.; Haynes, N.M.; Smyth, M.J.; Kroemer, G.; Zitvogel, L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med 2009, 15, 1170–1178. [Google Scholar]



©2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/)
Share and Cite
Chen, G.; Pedra, J.H.F. The Inflammasome in Host Defense. Sensors 2010, 10, 97-111. https://doi.org/10.3390/s100100097
Chen G, Pedra JHF. The Inflammasome in Host Defense. Sensors. 2010; 10(1):97-111. https://doi.org/10.3390/s100100097
Chicago/Turabian StyleChen, Gang, and Joao H.F. Pedra. 2010. "The Inflammasome in Host Defense" Sensors 10, no. 1: 97-111. https://doi.org/10.3390/s100100097
APA StyleChen, G., & Pedra, J. H. F. (2010). The Inflammasome in Host Defense. Sensors, 10(1), 97-111. https://doi.org/10.3390/s100100097