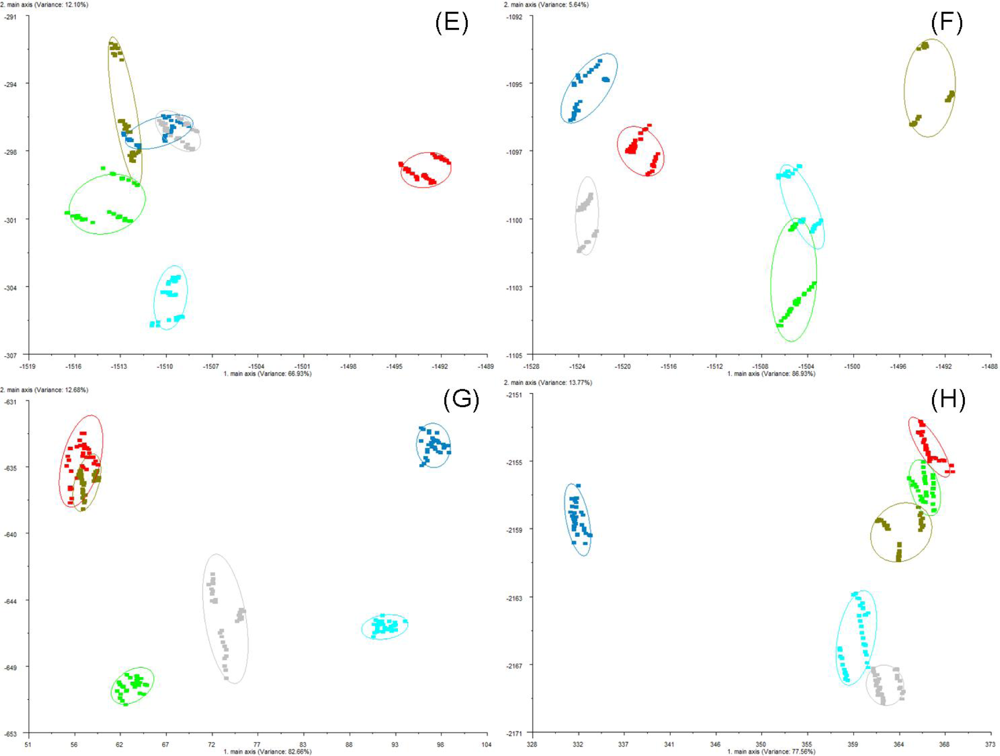
The AromaScan 32S is a conducting polymer electronic nose that contains an organic matrix-coated polymer-type 32-sensor array, designed for general use applications with 15 v across sensor paths. The sensor-array response to different VOCs was previously tested [
4]. Sensors responses are measured as a percentage of electrical resistance changes to current flow in the sensors relative to baseline resistance (%ΔR/R
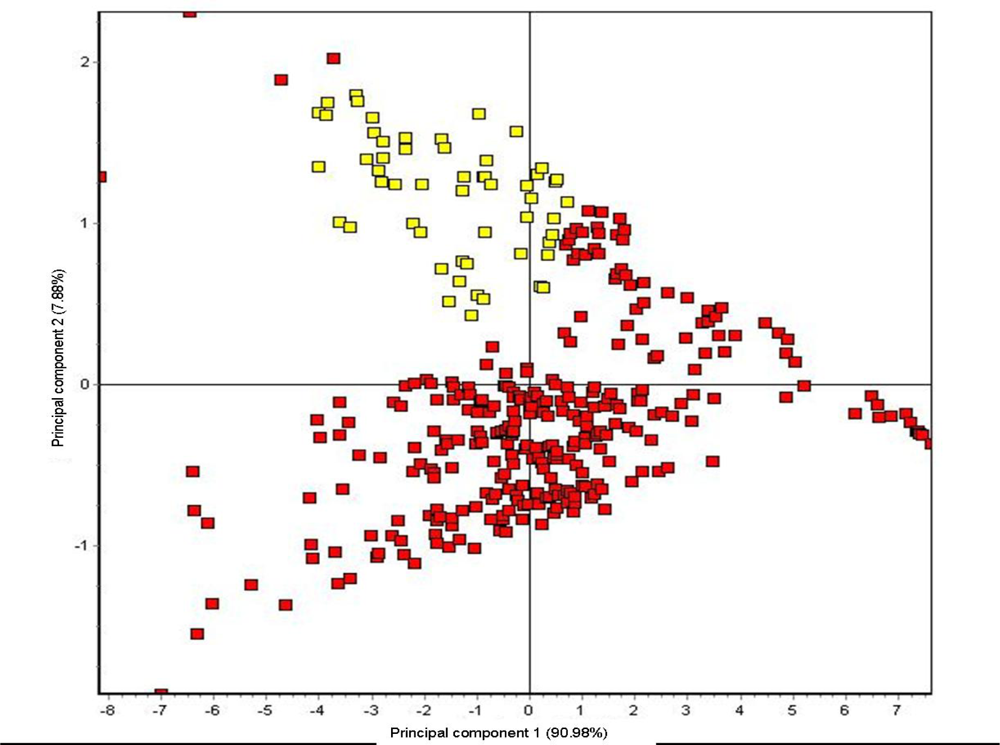
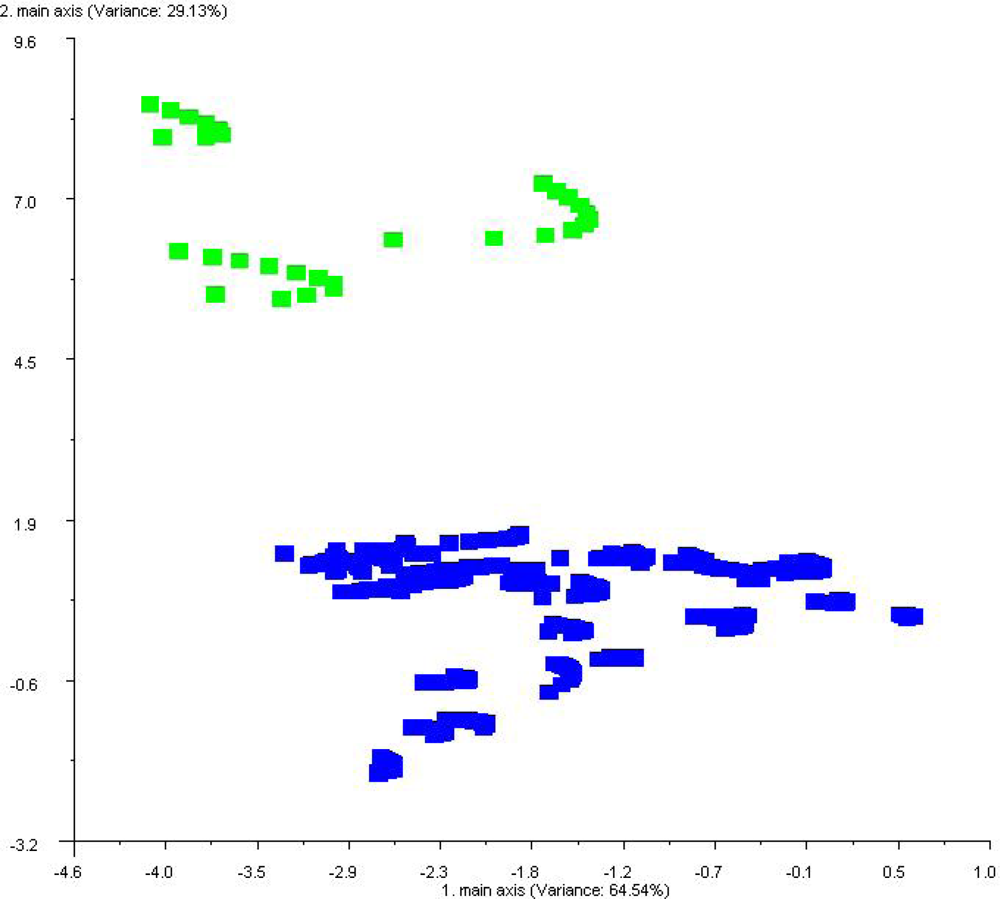
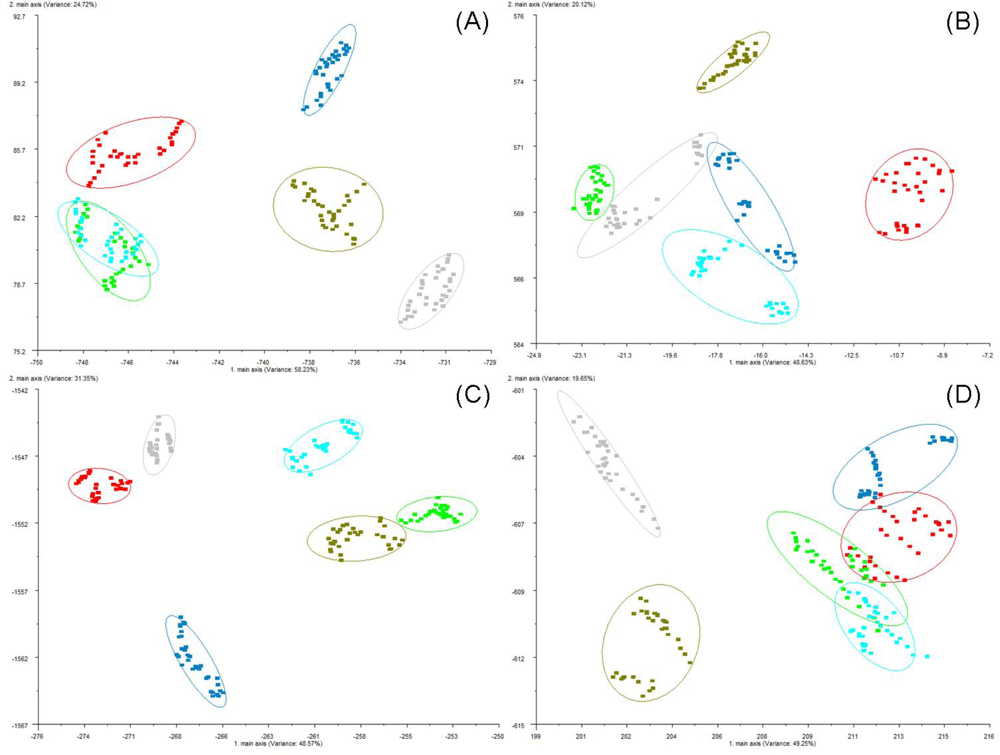
base). The sorption of headspace volatiles, composed of specific VOC mixtures, to the conducting polymer sensor surfaces induce a change in the electrical resistance to current flow which is detected and measured to produce the sensor-array output. Sensor responses varied with the type of plastic polymer used in the sensor matrix coating, produced by electropolymerization of either polypyrrole, polyanaline or polythiophene derivatives, which have been modified with ring-substitutions of different functional groups and with the addition of different types of metal ions to the polymer matrix in order to improve and modulate sensor response. All measurements were statistically compared using normalized sensor outputs from the sensor array. The conducting polymer analysis (CPA) methods used with this instrument employ application-specific reference libraries for aroma pattern recognition and neural-net training algorithms.
2.1.1. Samples Preparation
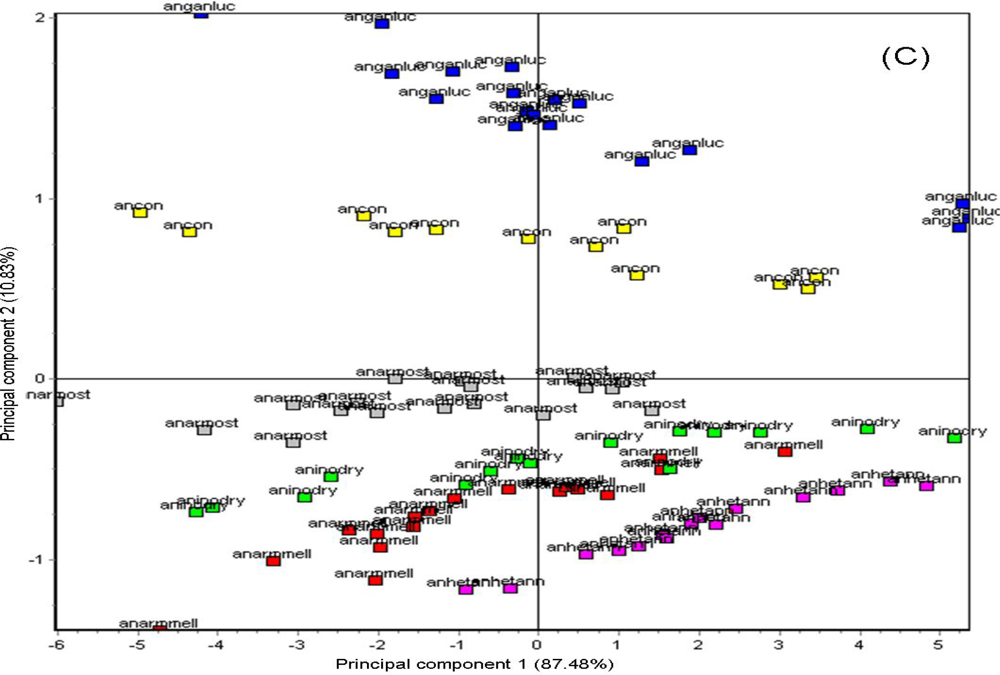
Headspace volatiles released from individual fungal cultures of twenty-four decay fungi, were analyzed individually (1–5 replications per sample type, based on availability) to test the capability of the AromaScan A32S to detect differences in fungal volatiles alone in pure culture. The wood decay fungi, including Amylostereum areolatum (Fr.) Boidin, Armillaria gallica Marxmüller and Romagnosi, Armillaria mellea (Vahl) P. Kumm, Armillaria ostoyae (Romagn.) Herink, Armillaria tabescens (Scop.) Emel, Cerrena unicolor (Bull.:Fr.) Murrill, Daedalea quercina (L.) Pers., Daldinia concentric (Bolton:Fr.) Ces. & De Not., Fomitopsis officinalis (Villars:Fr.) Bondartsev & Singer, Ganoderma lucidum (Curtis) P. Karst, Gloeophyllum sepiarium (Wulfen:Fr) P. Karst., Hericium erinaceus (Bull.:Fr.) Pers., Heterobasidion annosum (Fr.) Bref., Inonotus dryadeus (Pers. ex Fr.) Murr, Laetiporus sulphureus (Bull.) Murrill, Oxyporus latemarginatus (Durieu & Mont.) Donk, Phanerochaete gigantea (Fr.:Fr.) S.S. Rattan et al., Phellinus pini (Brot.) Bondartsev & Singer, Pleurotus ostreatus (Jacq.:Fr.) P. Kumm., Postia caesia (Shrad.:Fr.) P. Karst., Spongipellis pachyodon (Pers.) Kotlaba & Pouzar, Stereum hirsutum, (Willd.) Pers., Wolfiporia cocos (F.A. Wolf) Ryvarden & R.L. Gilbertson, and Xylaria polymorpha (Pers.:Fr.) Grev. were grown first on pure 4.5% Difco Malt agar (MA) culture medium 224200 (Becton Dickinson & Co., Sparks, MD, USA) in 100 × 15 mm-diameter plastic Petri dishes (Fisher Scientific, Pittsburgh, PA, USA) to make sure the cultures were pure without microbial contaminants. Pure axenic fungal culture samples for e-nose analysis were produced by transferring a single 5-mm plug of mycelium from a pure culture on Petri dishes into a 14.8 mL glass vials (21 × 70 mm) with screw-cap lids (Fisher Scientific), containing approximately 7 mL of the same MA medium, in order to allow the culture to grow and cover the 30°-slant agar surface over one to three weeks. The vials were capped, sealed and incubated at 25 °C ± 1 in the dark prior to analysis.
Wood samples from healthy trees were selected as controls for determinations of aroma signatures in the absence of decay. Tree bole sections 1–3 m long were collected from the trunks of nine tree species, including Populus deltoides Bartr. Ex Marshall, Fraxinus pennsylvanica Marsh., Liquidambar styraciflua L., Quercus nuttallii Palm., Platanus occidentalis L., Quercus lyrata Walt., Thuja occidentalis L., Pinus taeda L. and Taxodium distichum L., that were selected from among the hardwood and conifer species most common in the lower Mississippi Delta urban and forest environment. After one healthy tree of each species was cut down, the bole sections were placed in a desiccation room for about 24 hours at 60 C, peeled to remove the bark, and finally cut with a circular saw into 100 × 15 × 15 mm wood blocks (chip parallelepipeds). A minimum of 90 wood blocks from each tree was cut for a total of 810 wood block samples, and labeled using a felt-tipped marker. The samples were then sterilized in an autoclave at 121 °C for 15–20 minutes, dried in a 105 °C oven, then frozen at −20 °C in long-term storage until analyzed with each corresponding e-nose. These samples were considered “healthy” reference samples of which five replicate runs were analyzed for eight healthy wood types including: Fraxinus pennsylvanica, Liquidambar styraciflua, Pinus taeda, Populus deltoides, Quercus lyrata, Quercus nuttallii, Taxodium distichum, and Thuja occidentalis. These same control samples and decayed wood samples (fungus-host combinations) listed below were used in analyses for all three electronic noses, not just for the AromaScan A32S.
The “decayed samples” were produced by artificial inoculation of healthy wood blocks. A minimum of 3–6 wood blocks belonging to each of nine woody species (
Fraxinus pennsylvanica;
Liquidambar styraciflua;
Populus deltoides;
Plantanus occidentalis;
Pinus taeda;
Quercus nuttallii;
Quercus lyrata;
Taxodium distichum; and
Thuja occidentalis) was inoculated separately with 11 different wood decay fungi, including:
Armillaria gallica,
Armillaria mellea,
Armillaria ostoyae,
Armillaria tabescens,
Daedalea quercina,
Ganoderma lucidum,
Heterobasidion annosum,
Inonotus dryadeus,
Laetiporus sulphureus,
Phellinus pini (Thore:Fr.) A. Ames, and
Stereum hirsutum. Wood blocks of different host woods (
Fraxinus pennsylvanica;
Liquidambar styraciflua;
Populus deltoides;
Plantanus occidentalis;
Pinus taeda;
Quercus nuttallii;
Quercus lyrata;
Taxodium distichum; and
Thuja occidentalis) were artificially inoculated by dipping them into inoculum of each fungus using the following procedures. All of the isolates were previously preserved in sterile H
2O as mycelial plugs, in the USDA Pathology Lab of the Southern Research Station of Stoneville, within 1.8 mL cryotubes (Nunc A/S, Roskilde, Denmark) stored in a 5 °C refrigerator [
16]. Two plugs of mycelium were transferred from the cryotubes to sterile Petri plates containing 4.5% Malt agar M9802 (Sigma-Aldrich, St. Louis, MO, USA). The substrate was previously sterilized for 40 min in an autoclave at 121 °C and 15 psi, then poured into 10 cm Petri plates and cooled down to room temperature on a sterile surface within a laminar flow hood (Nuare Laminar Flow Products, Plymouth, MN, USA). Each of the two mycelium plugs was spatially placed far apart on the same Petri dish in order to obtain the widest culture margin possible between the fungal colonies. Every Petri dish was firmly sealed with Parafilm “M” (Pechiney Plastic Packaging, Chicago, IL, USA). In order to obtain pure cultures and to prevent further bacterial proliferation, all the isolates were transferred a second time to new sterile dishes containing 4.5% Malt agar medium. Eight mycelial plugs from each Petri dish culture were transferred into a 250 mL Pyrex
® sterile glass flask with 150 mL of 3% Malt Extract (Merck KGaA, Darmstadt, Germany) sterile broth. The flasks were then plugged with sterile cotton and gently shaken in order to distribute the plugs in the flasks. After one to three weeks, a large mass of mycelium was formed into the flasks. A hand-held stainless steel mixer (T10 basic Ultra-Turrax
®, IKA
® Werke GmbH & Co. KG, Staufen, Germany) with previously autoclaved blades was used to macerate the mycelial mass within the liquid culture. Inoculated wood blocks were incubated at 25 °C ± 1 in the dark for 12 months resting on 4.5% Malt agar substrate. After incubation, they were removed from the tubes, rinsed with tap water in order to remove every visible trace of fungal mycelium, blotted on tissue paper, wrapped in aluminum sheets, and stored at 5 °C.
2.1.2. Pre-Run Procedures and Data Collection
Healthy samples that became desiccated during storage were rehydrated by soaking in sterile distilled water for 15 minutes, followed by blotting on tissue paper to remove excess free moisture immediately prior to analysis. Healthy, decayed and pure culture samples were placed into a 500-ml Pyrex glass Laboratory storage sampling-bottle 1395 (Corning Inc., Corning, NY, USA), fitted with reference air, sampling, and exhaust ports on a polypropylene bottle cap, and firmly sealed to let the volatiles build headspace and equilibrate for 30 minutes prior to each run. The sampling bottle was held in the sampling chamber within the instrument at a constant air temperature of 25 °C ± 0.5 for the entire run period.
All pre-run procedures, instrument configurations, run parameters, data-acquisition settings, and run schedules followed the method specifications used by Wilson
et al. [
4]. Pre-run tests were performed to determine sample air relative humidity compared with that of reference air. Reference air was set at 4% relative humidity and adjusted to within 2% below sample air at 25 °C. The temperature of the sensor array was maintained at a constant 30 °C. Reference air was preconditioned by passing room air sequentially through a carbon filter with activated charcoal 05-685B (Fisher Scientific), silica gel beads (EMD Chemicals Inc., Gibbstown, NJ, USA), inline pump-protection filter 0500-0006 (Osmetech Inc., Wobum, MA, USA) and glass microfiber Hepa-Cap™ 36 filter (Whatman Inc., Clifton, NJ, USA) to remove organic compounds, moisture, particulates, and microbes, respectively. The flow rate (vacuum) of sample air at the sampling port was maintained at −700 ml/min using a calibrated ADM 3000 flow meter (Agilent Technologies, Wilmington, DE, USA). Sensors were purged between runs using a noncommercial 2% v/v isopropanol wash solution. The instrument was interfaced with a personal computer via a RS232 cable and controlled with AromaScan 3.51 software (Osmetech Inc.).
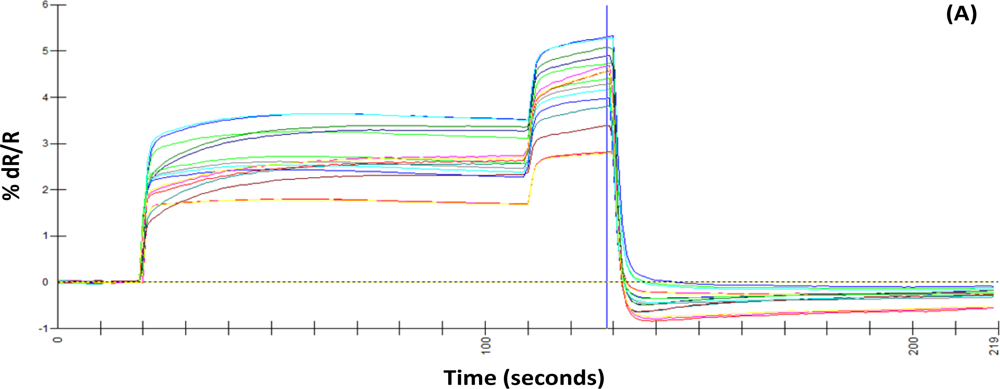
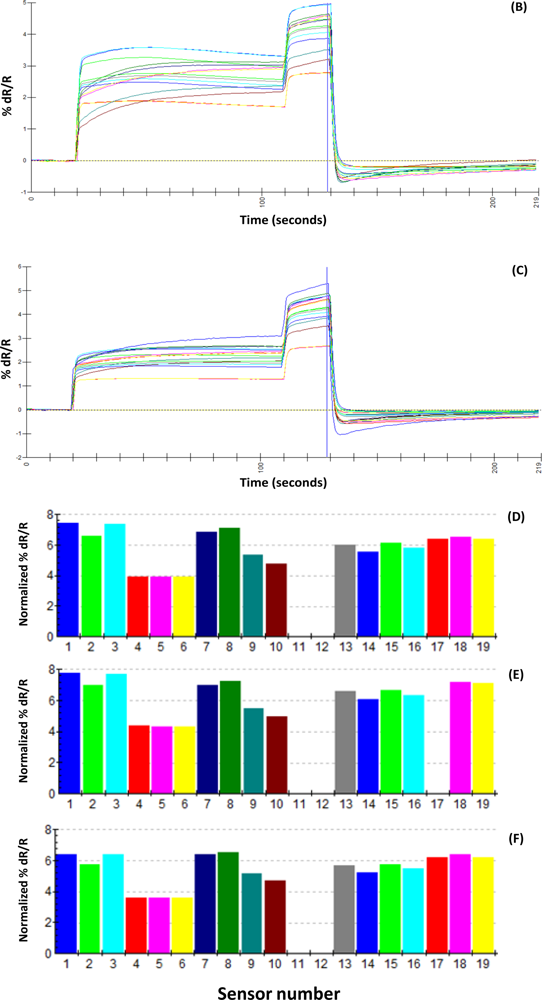
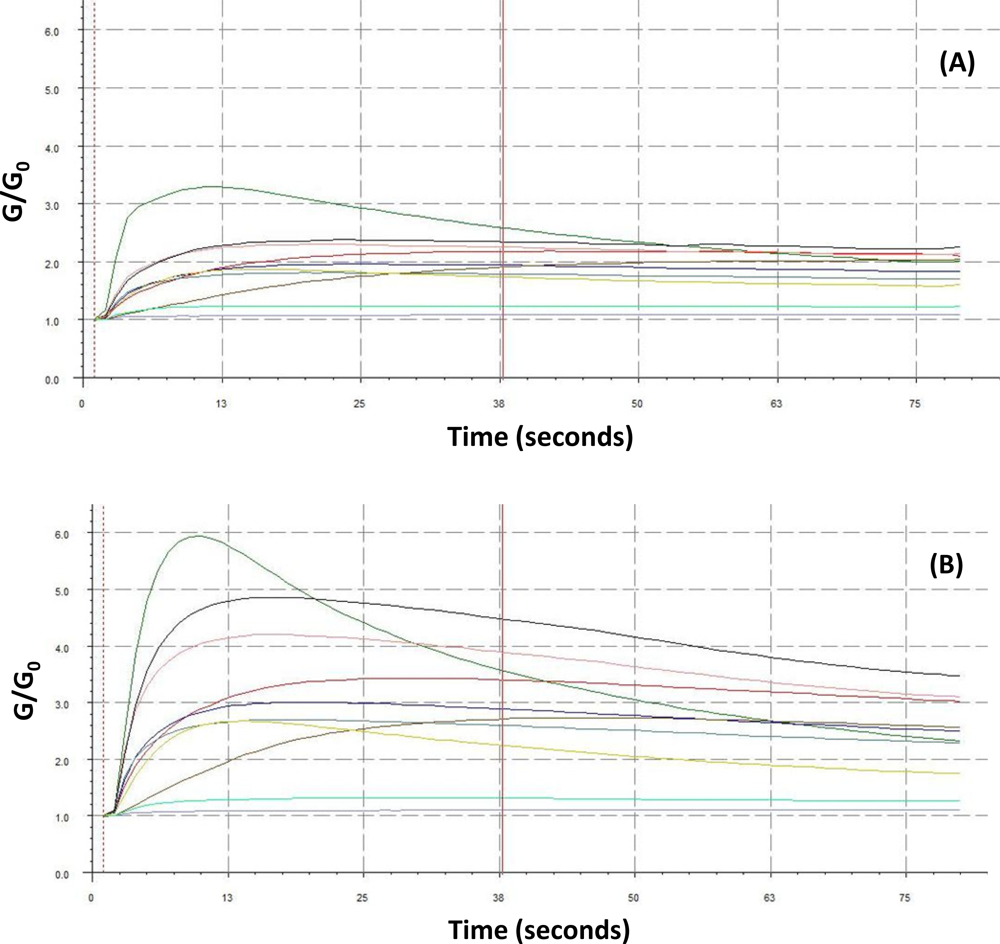
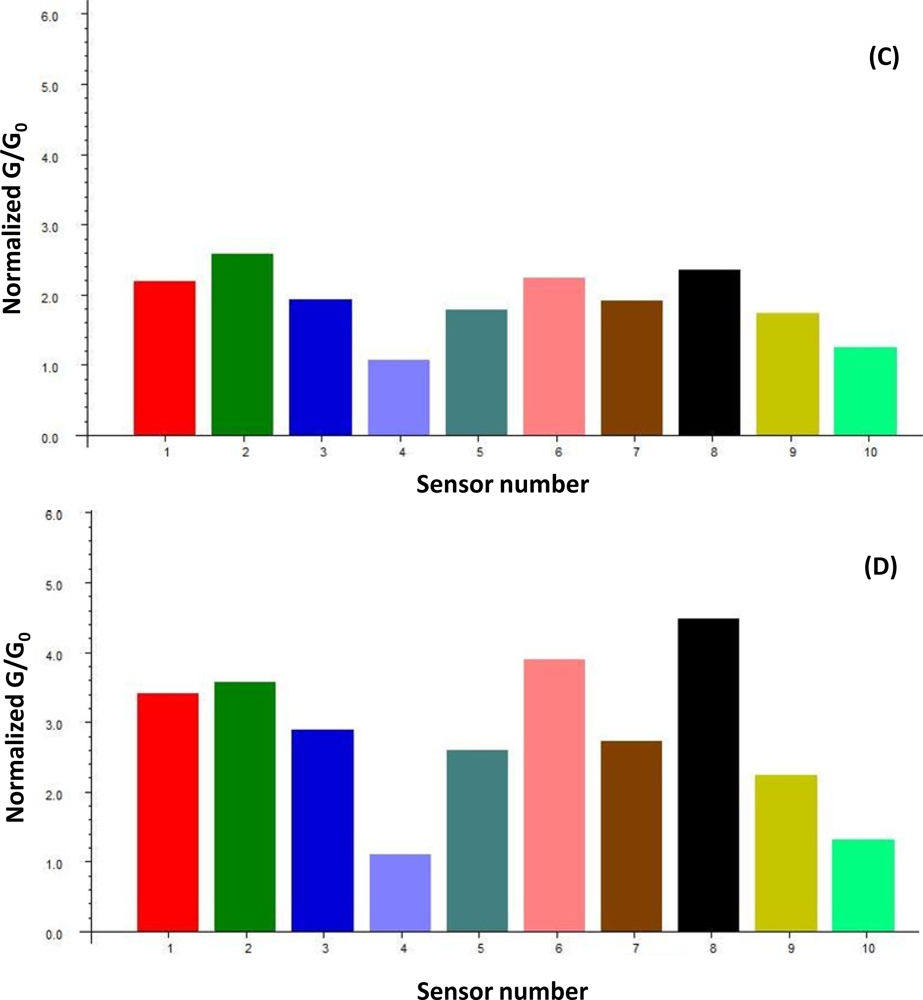
A uniform run schedule was used as following: reference air, 20 s; sampling time, 90 s; wash, 20 s; reference air, 90 s; a 2 min reference air purge followed by a 30 min equilibration period between the runs. Data from the sensor array were collected at 1 s intervals, and a conventional 20-s sampling interval (between 85 and 105 sec into the run) near the end of the sampling segment was used [
4].
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}