Raman Spectroscopy and Related Techniques in Biomedicine

Abstract
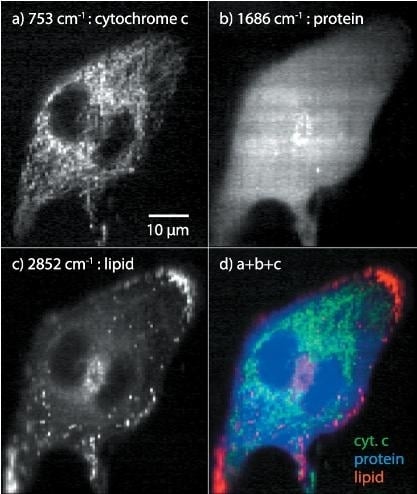
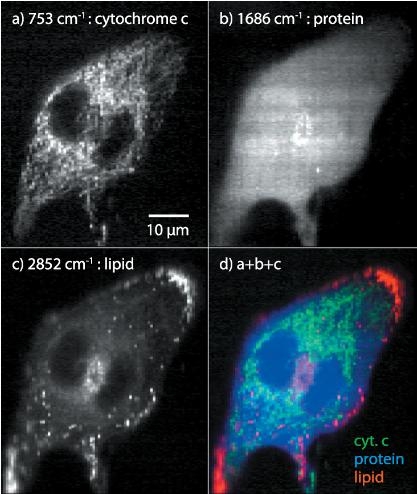
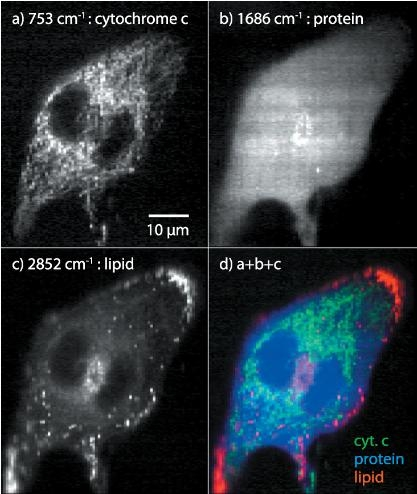
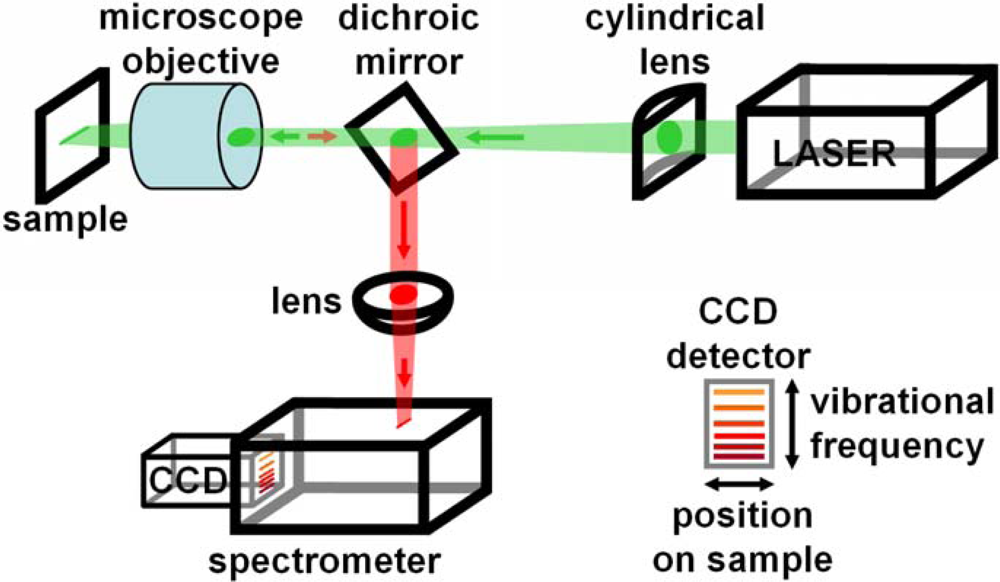
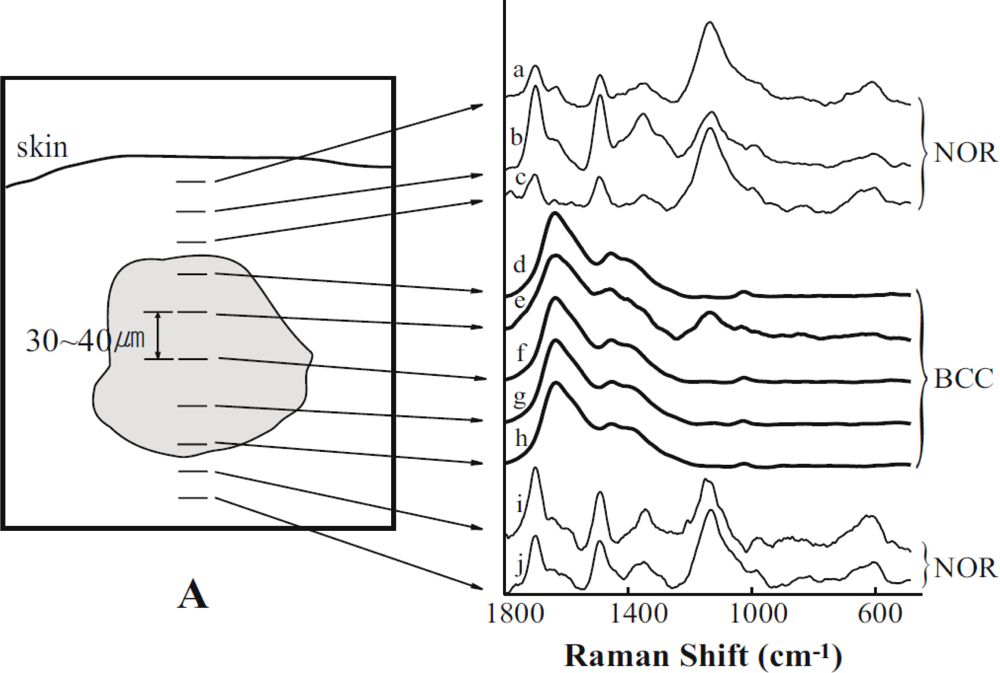
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. In Vitro Spectroscopy and Microscopy
3. In Vivo Spectroscopy
4. Statistical Analysis
5. Conclusions
Acknowledgments
References and Notes
- Yang, W.; Xiao, X.; Tan, J.; Cai, Q. In situ evaluation of breast cancer cell growth with 3D ATR-FTIR spectroscopy. Vib. Spectros 2009, 49, 64–67. [Google Scholar]
- Tobin, M.; Chesters, M.; Chalmers, J.; Rutten, F.; Fisher, S.; Symonds, I.; Hitchcock, A.; Allibone, R.; Dias-Gunasekara, S. Infrared microscopy of epithelial cancer cells in whole tissues and in tissue culture, using synchrotron radiation. Faraday Discus 2004, 126, 27–39. [Google Scholar]
- De Gelder, J.; De Gussem, K.; Vandenabeele, P.; Moens, L. Reference database of Raman spectra of biological molecules. J. Raman Spectrosc 2007, 38, 1133–1147. [Google Scholar]
- Liu, Y.; Sonek, G.; Berns, M.; Tromberg, B. Physiological monitoring of optically trapped cells: assessing the effects of confinement by 1064-nm laser tweezers using microfluorometry. Biophys. J 1996, 71, 2158–2167. [Google Scholar]
- Puppels, G.; Olminkhof, J.; Segers-Nolten, G.; Otto, C.; de Mul, F.; Greve, J. Laser irradiation and Raman spectroscopy of single living cells and chromosomes: sample degradation occurs with 514.5 nm but not with 660 nm laser light. Exp. Cell. Res 1991, 95, 361–367. [Google Scholar]
- Notingher, I.; Verrier, S.; Romanska, H.; Bishop, A.; Polak, J.; Hench, L. In situ characterisation of living cells by Raman spectroscopy. Spectroscopy 2002, 16, 43–51. [Google Scholar]
- Krafft, C.; Dietzek, B.; Popp, J. Raman and CARS microspectroscopy of cells and tissues. Analyst 2009, 134, 1046–1057. [Google Scholar]
- Ogawa, M.; Harada, Y.; Yamaoka, Y.; Fujita, K.; Takamatsu, T. Tissue imaging of myocardial infarct regions by a slit-scanning Raman microscope. Proc. SPIE 2009, 7169, 71690H. [Google Scholar]
- Hamada, K.; Fujita, K.; Smith, N.; Kobayashi, M.; Inouye, Y.; Kawata, S. Raman microscopy for dynamic molecular imaging of living cells. J Biomed. Opt 2008, 13, 044027. [Google Scholar]
- Kneipp, K.; Wang, Y.; Kneipp, H.; Perelman, L.; Itzkan, I.; Dasari, R.; Feld, M. Single molecule detection using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett 1997, 78, 1667–1670. [Google Scholar]
- Zavaleta, C.; Smith, B.; Walton, I.; Doering, W.; Davis, G.; Shojaei, B.; Natan, M.; Gambhir, S. Multiplexed imaging of surface enhanced Raman scattering nanotags in living mice using non-invasive Raman spectroscopy. PNAS 2009, 106, 13511–13516. [Google Scholar]
- Elfick, A.; Downes, A.; Mouras, R. Development of tip-enhanced optical spectroscopy for biological applications: a review. Anal. Bioanal. Chem 2010, 396, 45–52. [Google Scholar]
- Downes, A.; Mouras, R.; Mari, M.; Elfick, A. Optimising tip-enhanced optical microscopy. J. Raman Spectrosc 2009, 40, 1355–1360. [Google Scholar]
- Hartschuh, A.; Qian, H.; Meixner, A.; Anderson, N.; Novotny, L. Nanoscale optical imaging of single-walled carbon nanotubes. J. Luminesc 2006, (119–120), 204–208. [Google Scholar]
- Downes, A.; Salter, D.; Elfick, A. Heating effects in tip enhanced optical microscopy. Opt. Express 2006, 14, 5216–5222. [Google Scholar]
- Soudamini, D.; Fu, C.; Kho, K.; Thoniyot, P.; Agarwal, A.; Olivo, M. Fluctuation in surface enhanced Raman scattering intensity due to plasmon related heating effect. Proc. SPIE 2009, 7394, 73940T. [Google Scholar]
- Maher, R.; Cohen, L.; Le Rub, E.; Etchegoin, P. A study of local heating of molecules under Surface Enhanced Raman Scattering (SERS) conditions using the anti-Stokes/Stokes ratio. Faraday Discuss 2006, 132, 77–83. [Google Scholar]
- Kho, K.; Shen, Z.; Lei, Z.; Watt, F.; Soo, K.; Olivo, M. Investigation into a surface plasmon related heating effect in surface enhanced Raman spectroscopy. Anal. Chem 2007, 79, 8870–8882. [Google Scholar]
- Viets, C.; Hill, W. Laser power effects in SERS spectroscopy at thin metal films. J. Phys. Chem. B 2001, 105, 6330–6336. [Google Scholar]
- Duncan, M.; Reintjes, J.; Manuccia, T. Scanning coherent anti-Stokes Raman microscope. Opt. Lett 1982, 7, 350–352. [Google Scholar]
- Zumbusch, A.; Holtom, G.; Xie, X. Three-dimensional vibrational imaging by coherent anti-Stokes Raman scattering. Phys. Rev. Lett 1999, 82, 4142–4145. [Google Scholar]
- Cheng, J.; Xie, X. Coherent anti-Stokes Raman scattering microscopy: instrumentation, theory and applications. J. Phys. Chem. B 2004, 108, 827–840. [Google Scholar]
- Downes, A.; Mouras, R.; Elfick, A. A versatile CARS microscope for biological imaging. J. Raman Spectrosc 2009, 40, 757–762. [Google Scholar]
- Cheng, J.; Jia, Y.; Zheng, G.; Xie, X. Laser scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys. J 2002, 83, 502–509. [Google Scholar]
- Evans, C.; Potma, E.; Puoris’haag, M.; Côté, D.; Lin, C.; Xie, X. Chemical imaging of tissue in vivo with video-rate coherent anti-Stokes Raman scattering microscopy. PNAS 2005, 102, 16807–16812. [Google Scholar]
- Potma, E.; Evans, C.; Xie, X. Heterodyne coherent anti-Stokes Raman scattering (CARS) imaging. Opt. Lett 2006, 31, 241–243. [Google Scholar]
- Freudiger, C.; Min, W.; Saar, B.; Lu, S.; Holtom, G.; He, C.; Tsai, J.; Kang, J.; Xie, X. Label-free biomedical imaging with high sensitivity by stimulated Raman scattering microscopy. Science 2008, 322, 1857–1861. [Google Scholar]
- Cheng, J.; Volkmer, A.; Book, L.; Xie, X. Multiplex coherent anti-Stokes Raman scattering microspectroscopy and study of lipid vesicles. J. Phys. Chem. B 2002, 106, 8493–8498. [Google Scholar]
- Müller, M.; Schins, J. Imaging the thermodynamic state of lipid membranes with multiplex CARS microscopy. J. Phys. Chem. B 2002, 106, 3715–3723. [Google Scholar]
- Kee, T.; Cicerone, M. Simple approach to one-laser, broadband coherent anti-Stokes Raman scattering microscopy. Opt. Lett 2004, 29, 2701–2703. [Google Scholar]
- Kano, H.; Hamaguchi, H. In-vivo multi-nonlinear optical imaging of a living cell using a supercontinuum light source generated from a photonic crystal fiber. Opt. Express 2006, 14, 2798–2804. [Google Scholar]
- Slipchenko, M.; Le, T.; Chen, H.; Cheng, J. Compound Raman microscopy for high-speed vibrational imaging and spectral analysis of lipid bodies. J. Phys. Chem. B 2009, 113, 7681–7686. [Google Scholar]
- Cui, M.; Bachler, B.; Ogilvie, J. Comparing coherent and spontaneous Raman scattering under biological imaging conditions. Opt. Lett 2009, 34, 773–775. [Google Scholar]
- Harz, M.; Rösch, P.; Popp, J. Vibrational spectroscopy—A powerful tool for the rapid identification of microbial cells at the single-cell level. Cytometry 2009, 75A, 104–113. [Google Scholar]
- Nelson, W.; Manoharan, R.; Sperry, J. UV resonance Raman studies of bacteria. Appl. Spectrosc. Rev 1992, 27, 67–124. [Google Scholar]
- Notingher, I. Raman spectroscopy cell-based biosensors. Sensors 2007, 7, 1343–1358. [Google Scholar]
- Krafft, C.; Steiner, G.; Beleites, C.; Salzer, R. Disease recognition by infrared and Raman spectroscopy. J. Biophoton 2009, 2, 13–28. [Google Scholar]
- Keller, M.; Kanter, E.; Mahadevan-Jansen, A. Raman spectroscopy for cancer diagnosis. Spectroscopy 2006, 21, 33–41. [Google Scholar]
- Segers, V.; Lee, R. Stem-cell therapy for cardiac disease. Nature 2008, 451, 937–942. [Google Scholar]
- Langer, R. Tissue engineering: Perspectives, challenges and future directions. Tissue Eng 2007, 13, 1–2. [Google Scholar]
- Downes, A.; Mouras, R.; Elfick, A. Optical spectroscopy for non-invasive monitoring of stem cell differentiation. J. Biomed. Biotech 2010. [Google Scholar] [CrossRef]
- Chan, J.; Lieu, D. Label-free biochemical characterization of stem cells using vibrational spectroscopy. J. Biophoton 2009, 2, 656–668. [Google Scholar]
- Hoffman, L.; Carpenter, M. Characterization and culture of human embryonic stem cells. Nature Biotech 2005, 23, 699–708. [Google Scholar]
- Nagano, K.; Yoshida, Y.; Isobe, T. Cell surface biomarkers of embryonic stem cells. Proteomics 2008, 8, 4025–4035. [Google Scholar]
- Carden, A.; Morris, M. Application of vibrational spectroscopy to the study of mineralized tissues (review). J. Biomed. Opt 2000, 5, 259–268. [Google Scholar]
- Ashkin, A. Acceleration and trapping of particles by radiation pressure. Phys. Rev. Lett 1970, 24, 156–159. [Google Scholar]
- Xie, C.; Dinno, M.; Li, Y. Near-infrared Raman spectroscopy of single optically trapped biological cells. Opt. Lett 2002, 27, 249–251. [Google Scholar]
- Creelya, C.; Singha, G.; Petrov, D. Dual wavelength optical tweezers for confocal Raman spectroscopy. Opt. Comm 2005, 245, 465–470. [Google Scholar]
- Cheng, J.X.; Book, L.; Xie, X. Polarization coherent anti-Stokes Raman scattering microscopy. Opt. Lett 2001, 26, 1341–1343. [Google Scholar]
- Fujita, K.; Smith, N. Label-free molecular imaging of living cells. Mol. Cells 2008, 26, 530–535. [Google Scholar]
- Cheng, J.X.; Jia, Y.; Zheng, G.; Xie, X. Laser-scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys. J 2002, 83, 502–509. [Google Scholar]
- Holtom, G.; Thrall, B.; Chin, B.; Wiley, H.; Colson, S. Achieving molecular selectivity in imaging using multiphoton Raman spectroscopy techniques. Traffic 2001, 2, 781–788. [Google Scholar]
- Wang, H.; Le, T.; Cheng, J.X. Label-free imaging of arterial cells and extracellular matrix using a multimodal CARS microscope. Opt. Comm 2008, 281, 1813–1822. [Google Scholar]
- Wang, H.; Fu, Y; Zickmund, P.; Shi, R.; Cheng, J. Coherent anti-stokes Raman scattering imaging of axonal myelin in live spinal tissues. Biophys J 2005, 89, 581–591. [Google Scholar]
- Le, T.; Langohr, I.; Locker, M.; Sturek, M.; Cheng, J.X. Label-free molecular imaging of atherosclerotic lesions using multimodal nonlinear optical microscopy. J. Biomed. Opt 2007, 12, 054007. [Google Scholar]
- Mouras, R.; Rischitor, G.; Downes, A.; Salter, D.; Elfick, A. Nonlinear optical microscopy for drug delivery monitoring and cancer tissue imaging. J. Raman Spectrosc 2010. (accepted). [Google Scholar]
- Caspers, P.; Lucassen, G.; Puppels, G. Combined in vivo confocal Raman spectroscopy and confocal microscopy of human skin. Biophys. J 2003, 1, 572–580. [Google Scholar]
- Wright, A.; Poland, S.; Girkin, J.; Freudiger, C.; Evans, C.; Xie, X. Adaptive optics for enhanced signal in CARS microscopy. Opt. Express 2007, 15, 18209–18219. [Google Scholar]
- Komachi, Y.; Katagiri, T.; Sato, H.; Tashiro, H. Improvement and analysis of a micro Raman probe. Appl. Opt 2009, 48, 1683–1696. [Google Scholar]
- Buschman, H.; Marple, E.; Wach, M.; Bennett, B.; Bakker Schut, T.; Bruining, H.; Bruschke, A.; van der Laarse, A.; Puppels, G. In vivo determination of the molecular composition of artery wall by intravascular Raman spectroscopy. Anal. Chem 2000, 72, 3771–3775. [Google Scholar]
- Motz, J.; Fitzmaurice, M.; Miller, A.; Gandhi, S.; Haka, A.; Galindo, L.; Dasari, R.; Kramer, J.; Feld, M. In vivo Raman spectral pathology of human atherosclerosis and vulnerable plaque. J. Biomed. Opt 2006, 11, 021003. [Google Scholar]
- Erckens, R.; Jongsma, F.; Wicksted, J.; Hendrikse, F.; March, W.; Motamedi, M. Raman spectroscopy in ophthalmology: from experimental tool to applications in vivo. Lasers Med. Sci 2001, 16, 236–252. [Google Scholar]
- Glenn, J.; Renwick Beattie, J.; Barrett, L.; Frizzell, N.; Thorpe, S.; Boulton, M.; McGarvey, J.; Stitt, A. Confocal Raman microscopy can quantify advanced glycation end product (AGE) modifications in Bruch’s membrane leading to accurate, nondestructive prediction of ocular aging. FASEB J 2007, 21, 3542–3552. [Google Scholar]
- Qian, X.; Peng, X.; Ansari, D.; Yin-Goen, O.; Chen, G.; Shin, D.; Yang, L.; Young, A.; Wang, M.; Nie, S. In vivo tumor targeting and spectroscopic detection with surface-enhanced Raman nanoparticle tags. Nature Biotech 2008, 26, 83–90. [Google Scholar]
- Sharaf, M.; Illman, D.; Kowalski, B. Chemometrics; John Wiley & Sons: New York, NY, USA, 1986; Volume 82. [Google Scholar]
- Santos, R.; Sidaoui, H.; Silveira, L.; Pasqualucci, C.; Pacheco, M. Classification system of Raman spectra using cluster analysis to diagnose coronary artery lesions. Instr. Sci. Technol 2009, 37, 327–344. [Google Scholar]
- Hartigan, J. Clustering Algorithms; John Wiley & Sons: New York, NY, USA, 1975. [Google Scholar]
- Hartigan, J.; Wong, M.J. Algorithm AS 136: A k-means clustering algorithm. Royal Stat. Soc. C 1979, 28, 100–108. [Google Scholar]
- Ward, J. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc 1963, 58, 236–244. [Google Scholar]












© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Downes, A.; Elfick, A. Raman Spectroscopy and Related Techniques in Biomedicine. Sensors 2010, 10, 1871-1889. https://doi.org/10.3390/s100301871
Downes A, Elfick A. Raman Spectroscopy and Related Techniques in Biomedicine. Sensors. 2010; 10(3):1871-1889. https://doi.org/10.3390/s100301871
Chicago/Turabian StyleDownes, Andrew, and Alistair Elfick. 2010. "Raman Spectroscopy and Related Techniques in Biomedicine" Sensors 10, no. 3: 1871-1889. https://doi.org/10.3390/s100301871
APA StyleDownes, A., & Elfick, A. (2010). Raman Spectroscopy and Related Techniques in Biomedicine. Sensors, 10(3), 1871-1889. https://doi.org/10.3390/s100301871