
Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences



Abstract
:
1. Introduction


2. Considerations and Limitations of FRET Based Biosensors

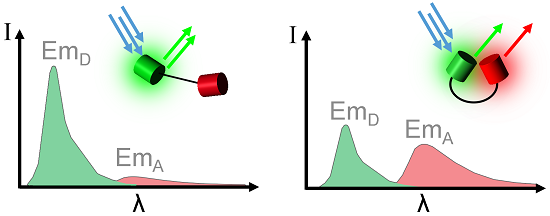{kind=link}
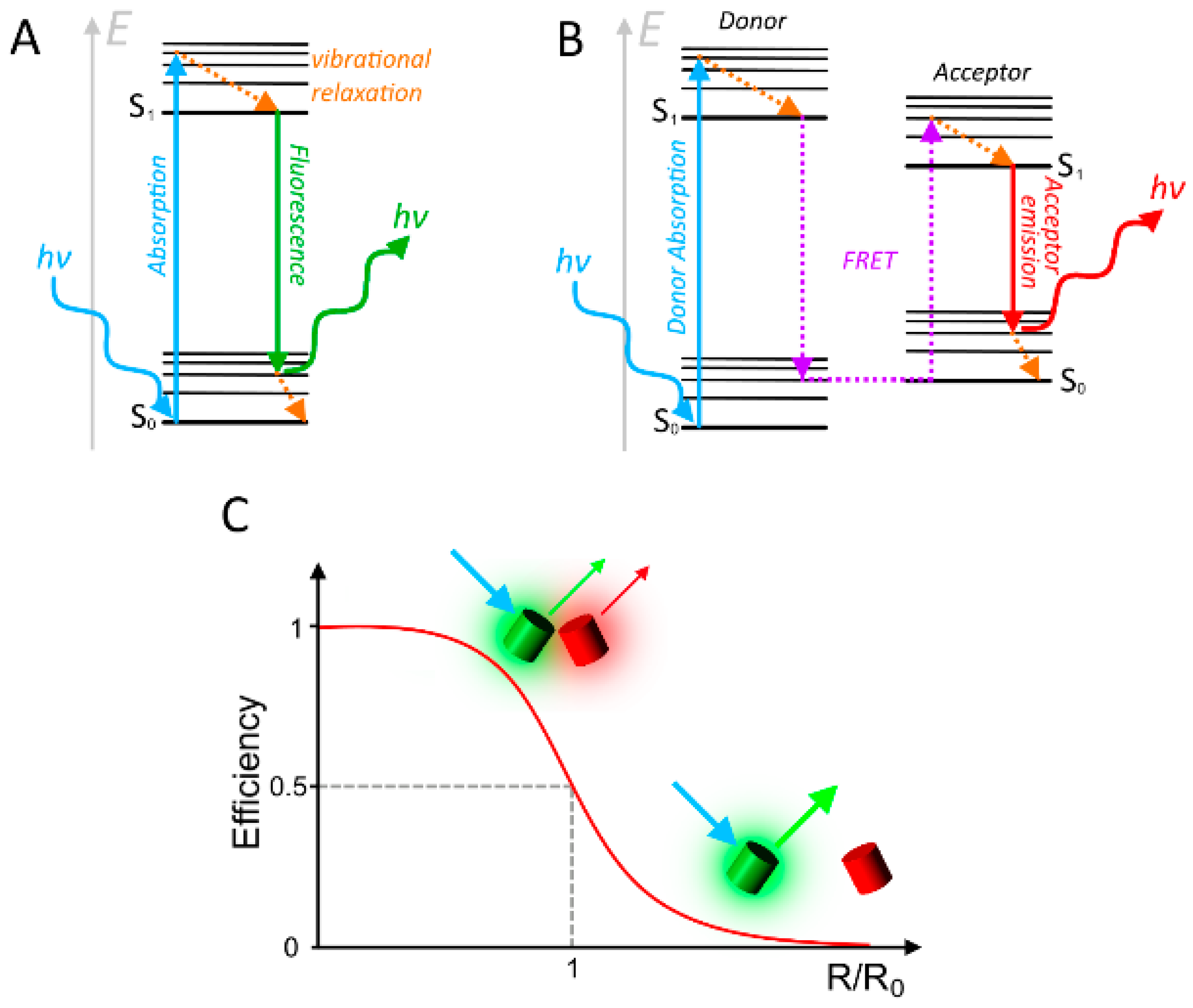{kind=link}
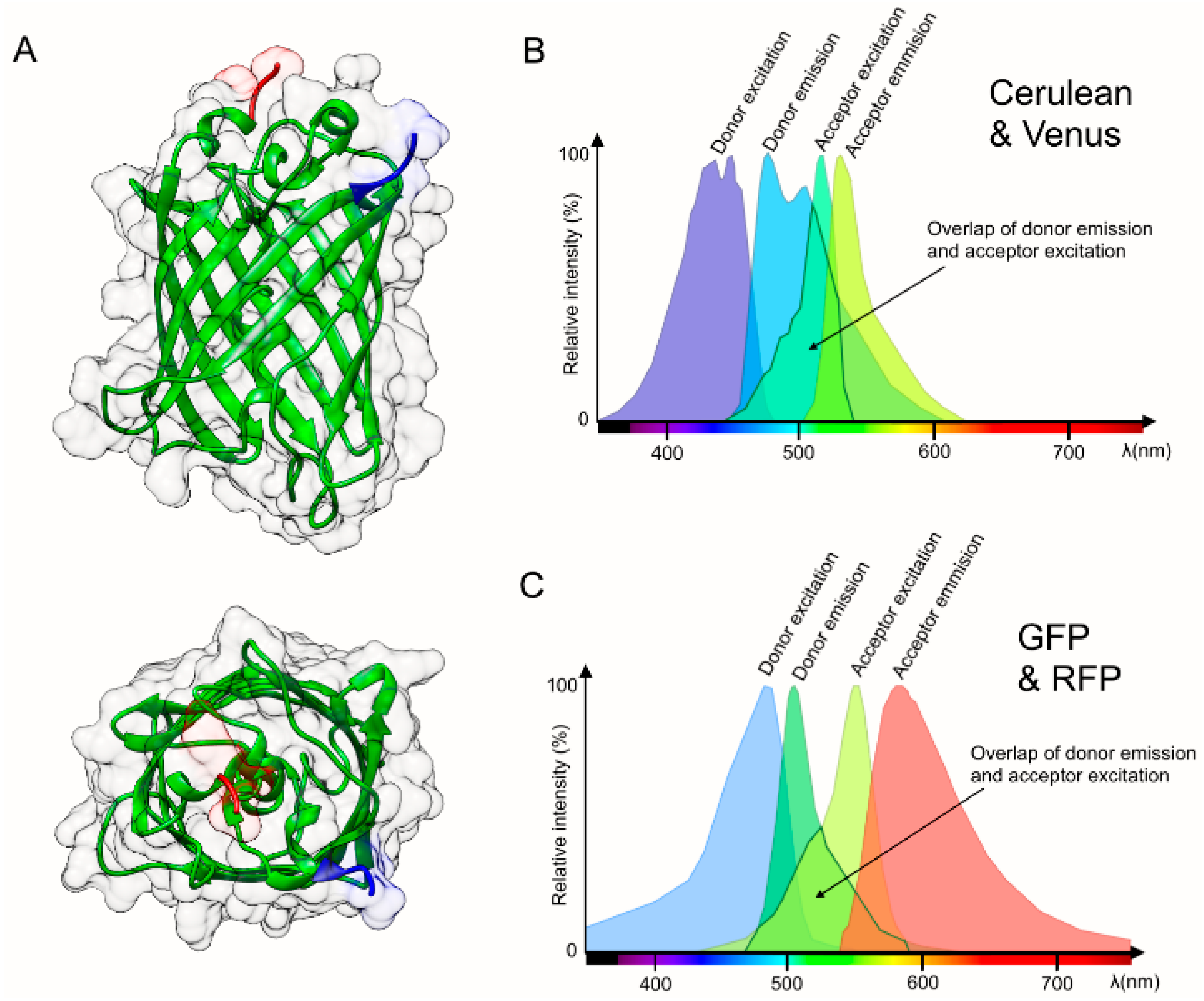{kind=link}
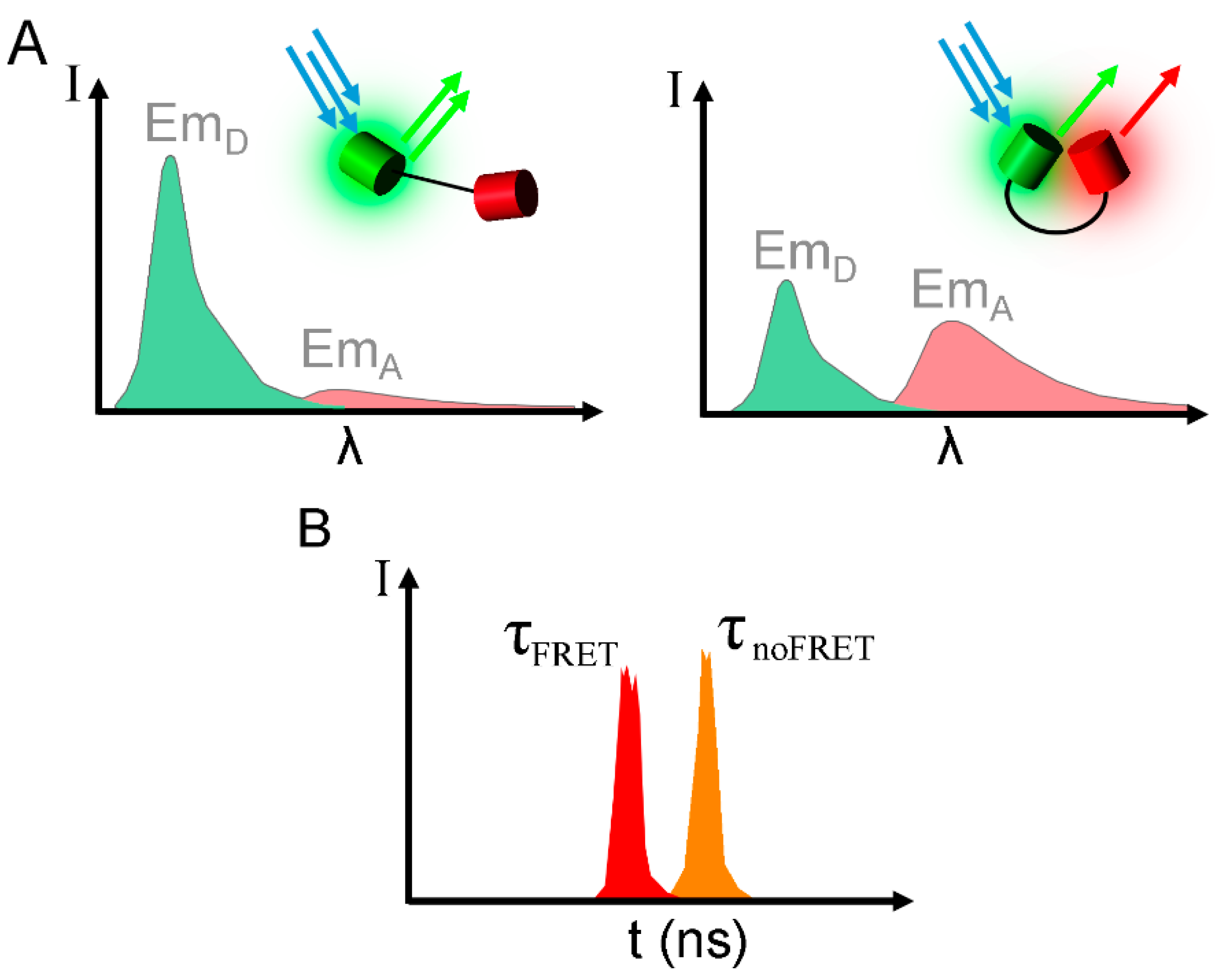{kind=link}
{kind=link}
{kind=link}
| Donor | Acceptor | Förster Radius R0 (nm) | Dynamic Range (nm) | Original Source |
|---|---|---|---|---|
| EBFP | EGFP | 4.1 | 2.1–6.2 | [17] |
| ECFP | EYFP | 4.9 | 2.5–7.3 | [17] |
| mCerulean | Venus | 5.4 | 2.7–8.1 | [18,19] |
| mCerulean | mCitrine | 5.4 | 2.7–8.1 | [18] |
| mTurquoise | mVenus | 5.7 | 2.9–8.6 | [19] |
| EGFP | EYFP | 5.6 | 2.8–8.4 | [17] |
| EGFP | DsRed | 4.7(5.8) | 2.4–7.1(2.9–8.7) | [20] |
| EGFP | mRFP1 | 4.7 | 2.4–7.1 | [21] |
| Clover | mRuby2 | 6.3 | 3.2–9.5 | [22] |
| Dronpa | mCherry | 5.6 | 2.8–8.4 | [12] |
| EYFP | DsRed | 4.9 | 2.5–7.4 | [17] |
| EYFP | mCherry | 5.7 | 2.9–8.6 | [23] |
2.1. FRET-Biosensor Design and Considerations
2.2. FRET vs. Single-FP Biosensors
3. Measurement and Evaluation of FRET
3.1. Fluorescence Intensity Based Methods

3.2. Fluorescence Lifetime Based Methods
3.3. Instrumentation for FRET-Measurements

4. Types of FRET Biosensors and Their Practical Applications
4.1. Cleavage Based FRET-Biosensors
| Target | Fluorophores | First Author, Year | Ref. | ||
|---|---|---|---|---|---|
| Cleavage | |||||
| Apoptosis | Caspase-3 | BFP, GFP | Xu, 1998 | [64] | |
| Caspase-3 | CFP, YFP | Tyas, 2000 | [65] | ||
| Caspase-8 & -9 | CFP, Venus | Takemoto, 2003 | [66] | ||
| Caspase-3 & -6 | CFP, YFP, mRFP | Wu, 2006 | [54] | ||
| Caspase-3 & -8 | CFP, YFP | Bozza, 2014 | [67] | ||
| Caspase-3 & -8 | seCFP, Venus, mRFP1 | Kominami, 2012 | [55] | ||
| Bid | CFP, YFP | Onuki, 2002 | [68] | ||
| Necroptosis | RIPK1 & RIPK3 | - | Sipieter, 2014 | [69] | |
| Autophagy | Atg4A & Atg4B | CFP, YFP | Li, 2012 | [70] | |
| ECM-remodelling | MT-MMP1 | Ypet, ECFP | Ouyang, 2008 | [71] | |
| MT-MMP1 | Ypet, ECFP | Lu, 2011 | [72] | ||
| MT-MMP1 | Orange2, Cherry | Eichorst, 2012 | [73] | ||
| Conformational Change | |||||
| Cell division | CyclinB1-Cdk1 | mCerulean, Ypet | Gavet, 2010 | [74] | |
| Signal transduction | AKT | ECFP, Ypet | Miura, 2014 | [75] | |
| AKT-PDK1 | CFP, YFP | Yoshizaki, 2007 | [76] | ||
| ERK | ECFP, Ypet | Kamioka, 2012 | [77] | ||
| Mechano Transduction | FAK | ECFP, Ypet | Seong, 2013 | [78] | |
| Src | ECFP, EYFP | Wang, 2005 | [79] | ||
| Metabolite Quantification | ATP | mseCFP, cp173-mVenus | Imamura, 2009 | [31] | |
| ATP | GFP, OFP | Vevea, 2013 | [80] | ||
| Glucose | EYFP, ECFP | Fehr, 2003 | [32] | ||
| Lactate | mTFP, Venus | San Martin, 2013 | [81] | ||
| Ca2+ | BFP, GFP or CFP, YFP | Miyawaki, 1997 | [43] | ||
| Drug Efficacy | BCR-ABL | M1Venus, ECFP | Mizutani, 2010 | [82] | |
| Src | ECFP, EYFP | Nobis, 2013 | [83] | ||
| T-cell interaction | ZAP-70 | CFP, YFP | Randriamampita, 2008 | [84] | |
| Lck | ECFP, EYFP | Paster, 2009 | [85] | ||
| Mechanical Force | |||||
| Focal Adhesion | Vinculin | mTFP1, Venus | Grashoff, 2010 | [86] | |
| Fluid Shear Stress | VE-cadherin PECAM-1 | mTFP1, Venus | Conway, 2013 | [87] | |
| E-cadherin | mTFP1, Venus | Borghi, 2012 | [88] | ||
| Changes in the Micro-Environment | |||||
| Oxygen & ROS | Oxygen | YFP, FbFP | Potzkei, 2012 | [15] | |
| ROS | ECFP, EYFP | Bernardini, 2015 | [89] | ||
| pH | pH | GFP, YFP | Awaji, 2001 | [90] | |
| pH | ECFP, EYFP | Urra, 2008 | [13] | ||

4.1.1. Apoptosis
4.1.2. Necroptosis
4.1.3. Autophagy
4.1.4. ECM-Remodelling
4.2. Conformational Change Based
4.2.1. Cell Division
4.2.2. Signal Transduction
4.2.3. Mechano-Transduction
4.2.4. Metabolite Quantification
4.2.5. Drug Efficacy
4.2.6. T-Cell Interaction
4.3. Mechanical Force Based
4.3.1. Focal Adhesion
4.3.2. Fluid Shear Stress
4.4. Sensors for Micro-Environmental Changes
4.4.1. Oxygen & ROS
4.4.2. pH
5. Conclusions/Outlook
Conflicts of Interest
References
- Stokes, G.G. On the change of refrangibility of light. Philos. Trans. R. Soc. Lond. 1852, 142, 463–562. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Annalen der Physik 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Lovell, J.F.; Chen, J.; Jarvi, M.T.; Cao, W.G.; Allen, A.D.; Liu, Y.; Tidwell, T.T.; Wilson, B.C.; Zheng, G. FRET quenching of photosensitizer singlet oxygen generation. J. Phys. Chem. B 2009, 113, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szollosi, J. Understanding FRET as a research tool for cellular studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Moss, L.G.; Phillips, G.N., Jr. The molecular structure of green fluorescent protein. Nat. Biotechnol. 1996, 14, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O. Discovery of green fluorescent protein (GFP) (nobel lecture). Angew. Chem. Int. Ed. Engl. 2009, 48, 5590–5602. [Google Scholar] [CrossRef] [PubMed]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the aequorea victoria green-fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Olenych, S.G.; Claxton, N.S.; Ottenberg, G.K.; Davidson, M.W. The fluorescent protein color palette. Curr Protoc Cell Biol. 2007. [Google Scholar] [CrossRef]
- Schmid, J.A.; Neumeier, H. Evolutions in science triggered by green fluorescent protein (GFP). Chembiochem 2005, 6, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.M.; Galliardt, H.; Schneider, J.; Barisas, B.G.; Seidel, T. Quantification of forster resonance energy transfer by monitoring sensitized emission in living plant cells. Front. Plant. Sci 2013, 4, 413. [Google Scholar] [CrossRef]
- Urra, J.; Sandoval, M.; Cornejo, I.; Barros, L.F.; Sepulveda, F.V.; Cid, L.P. A genetically encoded ratiometric sensor to measure extracellular ph in microdomains bounded by basolateral membranes of epithelial cells. Pflugers Arch. 2008, 457, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Navaratnam, D.; Santos-Sacchi, J. A genetically-encoded YFP sensor with enhanced chloride sensitivity, photostability and reduced ph interference demonstrates augmented transmembrane chloride movement by gerbil prestin (slc26a5). PLoS ONE 2014, 9, e99095. [Google Scholar] [CrossRef] [PubMed]
- Potzkei, J.; Kunze, M.; Drepper, T.; Gensch, T.; Jaeger, K.E.; Buchs, J. Real-time determination of intracellular oxygen in bacteria using a genetically encoded FRET-based biosensor. BMC Biol. 2012, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Piston, D.W.; Barisas, B.G. Forster distances between green fluorescent protein pairs. Anal. Biochem. 2000, 284, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Springer, G.; Segawa, K.; Zipfel, W.R.; Piston, D.W. Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins. Microsc. Microanal. 2006, 12, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, M.L.; Kremers, G.J.; Kraft, C.A.; Ray, K.; Cranfill, P.J.; Wilson, K.A.; Day, R.N.; Wachter, R.M.; Davidson, M.W.; Rizzo, M.A. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS ONE 2011, 6, e17896. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.G.; Moon, D.L.; Yue, D.T. Dsred as a potential FRET partner with CFP and GFP. Biophys. J. 2003, 85, 599–611. [Google Scholar] [CrossRef]
- Peter, M.; Ameer-Beg, S.M.; Hughes, M.K.; Keppler, M.D.; Prag, S.; Marsh, M.; Vojnovic, B.; Ng, T. Multiphoton-flim quantification of the eGFP-MRFP1 FRET pair for localization of membrane receptor-kinase interactions. Biophys. J. 2005, 88, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Akrap, N.; Seidel, T.; Barisas, B.G. Forster distances for fluorescence resonant energy transfer between mcherry and other visible fluorescent proteins. Anal. Biochem. 2010, 402, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Malkani, N.; Schmid, J.A. Some secrets of fluorescent proteins: Distinct bleaching in various mounting fluids and photoactivation of cyan fluorescent proteins at YFP-excitation. PLoS ONE 2011, 6, e18586. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Springer, G.H.; Granada, B.; Piston, D.W. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 2004, 22, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; van Weeren, L.; Hink, M.A.; Vischer, N.O.; Jalink, K.; Gadella, T.W., Jr. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods 2010, 7, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Matz, M.V.; Fradkov, A.F.; Labas, Y.A.; Savitsky, A.P.; Zaraisky, A.G.; Markelov, M.L.; Lukyanov, S.A. Fluorescent proteins from nonbioluminescent anthozoa species. Nat. Biotechnol. 1999, 17, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W.; et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 2007, 4, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Kredel, S.; Oswald, F.; Nienhaus, K.; Deuschle, K.; Rocker, C.; Wolff, M.; Heilker, R.; Nienhaus, G.U.; Wiedenmann, J. Mruby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS ONE 2009, 4, e4391. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, A.; Wittmann, T. Fluorescence live cell imaging. Methods Cell Biol. 2014, 123, 77–94. [Google Scholar] [PubMed]
- Imamura, H.; Nhat, K.P.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef] [PubMed]
- Fehr, M.; Lalonde, S.; Lager, I.; Wolff, M.W.; Frommer, W.B. In vivo imaging of the dynamics of glucose uptake in the cytosol of cos-7 cells by fluorescent nanosensors. J. Biol. Chem. 2003, 278, 19127–19133. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, S.; Looger, L.L.; Micheva, K.D.; Reimer, R.J.; Smith, S.J.; Frommer, W.B. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc. Natl. Acad. Sci. USA 2005, 102, 8740–8745. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, K.; Chaudhuri, B.; Okumoto, S.; Lager, I.; Lalonde, S.; Frommer, W.B. Rapid metabolism of glucose detected with FRET glucose nanosensors in epidermal cells and intact roots of arabidopsis rna-silencing mutants. Plant. Cell 2006, 18, 2314–2325. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Bernard, C.S.; Hubert, P.; My, L.; Zhang, C.C. Fluorescence resonance energy transfer based on interaction of PII and PIPX proteins provides a robust and specific biosensor for 2-oxoglutarate, a central metabolite and a signaling molecule. FEBS J. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hires, S.A.; Zhu, Y.; Tsien, R.Y. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc. Natl. Acad. Sci. USA 2008, 105, 4411–4416. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and gtpases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Yamada, S.; Tominaga, T.; Ichikawa, M.; Miyawaki, A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 10554–10559. [Google Scholar] [CrossRef] [PubMed]
- Van der Krogt, G.N.; Ogink, J.; Ponsioen, B.; Jalink, K. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: Application to the epac camp sensor as an example. PLoS ONE 2008, 3, e1916. [Google Scholar] [CrossRef] [PubMed]
- Vinkenborg, J.L.; Evers, T.H.; Reulen, S.W.; Meijer, E.W.; Merkx, M. Enhanced sensitivity of FRET-based protease sensors by redesign of the GFP dimerization interface. Chembiochem 2007, 8, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Kotera, I.; Iwasaki, T.; Imamura, H.; Noji, H.; Nagai, T. Reversible dimerization of aequorea victoria fluorescent proteins increases the dynamic range of FRET-based indicators. ACS Chem. Biol. 2010, 5, 215–222. [Google Scholar] [CrossRef]
- Nguyen, A.W.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [PubMed]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [PubMed]
- Frommer, W.B.; Davidson, M.W.; Campbell, R.E. Genetically encoded biosensors based on engineered fluorescent proteins. Chem. Soc. Rev. 2009, 38, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Rivera, J.D.; Guilbe, M.M.; Malave, E.C.; Hernandez, H.H.; Tian, L.; Hires, S.A.; Marvin, J.S.; Looger, L.L.; Schreiter, E.R. Crystal structures of the gcamp calcium sensor reveal the mechanism of fluorescence signal change and aid rational design. J. Biol. Chem. 2009, 284, 6455–6464. [Google Scholar] [CrossRef] [PubMed]
- Bonnot, A.; Guiot, E.; Hepp, R.; Cavellini, L.; Tricoire, L.; Lambolez, B. Single-fluorophore biosensors based on conformation-sensitive GFP variants. FASEB J. 2014, 28, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Puhl, H.L., 3rd; Koushik, S.V.; Vogel, S.S.; Ikeda, S.R. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys. J. 2006, 91, L39–L41. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Rietdorf, J.; Pepperkok, R. Spectral imaging and its applications in live cell microscopy. FEBS Lett. 2003, 546, 87–92. [Google Scholar] [CrossRef]
- Su, T.; Pan, S.; Luo, Q.; Zhang, Z. Monitoring of dual bio-molecular events using FRET biosensors based on mtagbfp/sfgfp and mvenus/mkokappa fluorescent protein pairs. Biosens. Bioelectron. 2013, 46, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Woehler, A. Simultaneous quantitative live cell imaging of multiple FRET-based biosensors. PLoS ONE 2013, 8, e61096. [Google Scholar] [CrossRef] [PubMed]
- Geissler, D.; Stufler, S.; Lohmannsroben, H.G.; Hildebrandt, N. Six-color time-resolved forster resonance energy transfer for ultrasensitive multiplexed biosensing. J. Am. Chem. Soc. 2013, 135, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Simone, J.; Hewgill, D.; Siegel, R.; Lipsky, P.E.; He, L. Measurement of two caspase activities simultaneously in living cells by a novel dual FRET fluorescent indicator probe. Cytometry A 2006, 69, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Kominami, K.; Nagai, T.; Sawasaki, T.; Tsujimura, Y.; Yashima, K.; Sunaga, Y.; Tsuchimochi, M.; Nishimura, J.; Chiba, K.; Nakabayashi, J.; et al. In vivo imaging of hierarchical spatiotemporal activation of caspase-8 during apoptosis. PLoS ONE 2012, 7, e50218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakowicz, J.R.; Szmacinski, H.; Nowaczyk, K.; Berndt, K.W.; Johnson, M. Fluorescence lifetime imaging. Anal. Biochem. 1992, 202, 316–330. [Google Scholar] [CrossRef]
- Hum, J.M.; Siegel, A.P.; Pavalko, F.M.; Day, R.N. Monitoring biosensor activity in living cells with fluorescence lifetime imaging microscopy. Int. J. Mol. Sci. 2012, 13, 14385–14400. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P. Advanced fluorescence microscopy techniques—FRAP, flip, flap, FRET and flim. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbe, E. Vii.—on the estimation of aperture in the microscope. J. R. Microsc. Soc. 1881, 1, 388–423. [Google Scholar] [CrossRef]
- Rinnenthal, J.L.; Bornchen, C.; Radbruch, H.; Andresen, V.; Mossakowski, A.; Siffrin, V.; Seelemann, T.; Spiecker, H.; Moll, I.; Herz, J.; et al. Parallelized TCSPC for dynamic intravital fluorescence lifetime imaging: Quantifying neuronal dysfunction in neuroinflammation. PLoS ONE 2013, 8, e60100. [Google Scholar] [CrossRef] [PubMed]
- Hinde, E.; Digman, M.A.; Welch, C.; Hahn, K.M.; Gratton, E. Biosensor forster resonance energy transfer detection by the phasor approach to fluorescence lifetime imaging microscopy. Microsc. Res. Tech. 2012, 75, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Fluorescence lifetime imaging—techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Jan Willem, B.; Antonie, J.W.G.V. Fluorescence lifetime imaging microscopy in life sciences. Meas. Sci. Technol. 2010, 21, 102002. [Google Scholar]
- Xu, X.; Gerard, A.L.; Huang, B.C.; Anderson, D.C.; Payan, D.G.; Luo, Y. Detection of programmed cell death using fluorescence energy transfer. Nucl. Acids Res. 1998, 26, 2034–2035. [Google Scholar] [CrossRef] [PubMed]
- Tyas, L.; Brophy, V.A.; Pope, A.; Rivett, A.J.; Tavare, J.M. Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Rep. 2000, 1, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Nagai, T.; Miyawaki, A.; Miura, M. Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effects. J. Cell Biol. 2003, 160, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Bozza, W.P.; Di, X.; Takeda, K.; Rivera Rosado, L.A.; Pariser, S.; Zhang, B. The use of a stably expressed FRET biosensor for determining the potency of cancer drugs. PLoS ONE 2014, 9, e107010. [Google Scholar] [CrossRef] [PubMed]
- Onuki, R.; Nagasaki, A.; Kawasaki, H.; Baba, T.; Uyeda, T.Q.; Taira, K. Confirmation by FRET in individual living cells of the absence of significant amyloid beta -mediated caspase 8 activation. Proc. Natl. Acad. Sci. USA 2002, 99, 14716–14721. [Google Scholar] [CrossRef] [PubMed]
- Sipieter, F.; Ladik, M.; Vandenabeele, P.; Riquet, F. Shining light on cell death processes—A novel biosensor for necroptosis, a newly described cell death program. Biotechnol J. 2014, 9, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, X.; Ye, Q.Z.; Vogt, A.; Yin, X.M. A high-throughput FRET-based assay for determination of ATG4 activity. Autophagy 2012, 8, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Lu, S.; Li, X.Y.; Xu, J.; Seong, J.; Giepmans, B.N.; Shyy, J.Y.; Weiss, S.J.; Wang, Y. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. J. Biol. Chem. 2008, 283, 17740–17748. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Eichorst, J.P.; Clegg, R.M.; Wang, Y. Red-shifted fluorescent proteins monitor enzymatic activity in live ht-1080 cells with fluorescence lifetime imaging microscopy (flim). J. Microsc. 2012, 248, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Progressive activation of cyclinb1-cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Matsuda, M.; Aoki, K. Development of a FRET biosensor with high specificity for akt. Cell. Struct. Funct. 2014, 39, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, H.; Mochizuki, N.; Gotoh, Y.; Matsuda, M. Akt-pdk1 complex mediates epidermal growth factor-induced membrane protrusion through RAL activation. Mol. Biol Cell 2007, 18, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Kamioka, Y.; Sumiyama, K.; Mizuno, R.; Sakai, Y.; Hirata, E.; Kiyokawa, E.; Matsuda, M. Live imaging of protein kinase activities in transgenic mice expressing FRET biosensors. Cell Struct. Funct. 2012, 37, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; Tajik, A.; Sun, J.; Guan, J.L.; Humphries, M.J.; Craig, S.E.; Shekaran, A.; Garcia, A.J.; Lu, S.; Lin, M.Z.; et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 19372–19377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Botvinick, E.L.; Zhao, Y.; Berns, M.W.; Usami, S.; Tsien, R.Y.; Chien, S. Visualizing the mechanical activation of SRC. Nature 2005, 434, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Vevea, J.D.; Wolken, D.M.; Swayne, T.C.; White, A.B.; Pon, L.A. Ratiometric biosensors that measure mitochondrial redox state and atp in living yeast cells. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- San Martin, A.; Ceballo, S.; Ruminot, I.; Lerchundi, R.; Frommer, W.B.; Barros, L.F. A genetically encoded FRET lactate sensor and its use to detect the warburg effect in single cancer cells. PLoS ONE 2013, 8, e57712. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Kondo, T.; Darmanin, S.; Tsuda, M.; Tanaka, S.; Tobiume, M.; Asaka, M.; Ohba, Y. A novel FRET-based biosensor for the measurement of bcr-abl activity and its response to drugs in living cells. Clin. Cancer Res. 2010, 16, 3964–3975. [Google Scholar] [CrossRef] [PubMed]
- Nobis, M.; McGhee, E.J.; Morton, J.P.; Schwarz, J.P.; Karim, S.A.; Quinn, J.; Edward, M.; Campbell, A.D.; McGarry, L.C.; Evans, T.R.; et al. Intravital flim-FRET imaging reveals dasatinib-induced spatial control of src in pancreatic cancer. Cancer Res. 2013, 73, 4674–4686. [Google Scholar] [CrossRef] [PubMed]
- Randriamampita, C.; Mouchacca, P.; Malissen, B.; Marguet, D.; Trautmann, A.; Lellouch, A.C. A novel zap-70 dependent FRET based biosensor reveals kinase activity at both the immunological synapse and the antisynapse. PLoS ONE 2008, 3, e1521. [Google Scholar] [CrossRef] [PubMed]
- Paster, W.; Paar, C.; Eckerstorfer, P.; Jakober, A.; Drbal, K.; Schutz, G.J.; Sonnleitner, A.; Stockinger, H. Genetically encoded forster resonance energy transfer sensors for the conformation of the src family kinase lck. J. Immunol. 2009, 182, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Grashoff, C.; Hoffman, B.D.; Brenner, M.D.; Zhou, R.; Parsons, M.; Yang, M.T.; McLean, M.A.; Sligar, S.G.; Chen, C.S.; Ha, T.; et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 2010, 466, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.E.; Breckenridge, M.T.; Hinde, E.; Gratton, E.; Chen, C.S.; Schwartz, M.A. Fluid shear stress on endothelial cells modulates mechanical tension across ve-cadherin and pecam-1. Curr. Biol. 2013, 23, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568–12573. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, A.; Brockmeier, U.; Metzen, E.; Berchner-Pfannschmidt, U.; Harde, E.; Acker-Palmer, A.; Papkovsky, D.; Acker, H.; Fandrey, J. Type i cell ros kinetics under hypoxia in the intact mouse carotid body ex vivo: A FRET-based study. Am. J. Physiol. Cell Physiol. 2015, 308, C61–C67. [Google Scholar] [CrossRef] [PubMed]
- Awaji, T.; Hirasawa, A.; Shirakawa, H.; Tsujimoto, G.; Miyazaki, S. Novel green fluorescent protein-based ratiometric indicators for monitoring ph in defined intracellular microdomains. Biochem. Biophys. Res. Commun. 2001, 289, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Means to an End: Apoptosis and Other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011. [Google Scholar]
- Miura, M. Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.B.; Green, D.R. Suicidal tendencies: Apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999, 274, 20049–20052. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(adp-ribose) polymerase by a proteinase with properties like ice. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Rehm, M.; Dussmann, H.; Janicke, R.U.; Tavare, J.M.; Kogel, D.; Prehn, J.H. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J. Biol. Chem. 2002, 277, 24506–24514. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ip, L.; Luo, H.; Chang, D.C.; Luo, K.Q. A high throughput drug screen based on fluorescence resonance energy transfer (FRET) for anticancer activity of compounds from herbal medicine. Br. J. Pharmacol. 2007, 150, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Fu, A.; Teoh, S.H.; Luo, K.Q. Application of a fluorescence resonance energy transfer (FRET)-based biosensor for detection of drug-induced apoptosis in a 3d breast tumor model. Biotechnol. Bioeng. 2015, 112, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Shinotsuka, N.; Nonomura, K.; Takemoto, K.; Kuida, K.; Yosida, H.; Miura, M. Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J. Cell. Biol. 2011, 195, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts). N. Eng. J. Med. 1980, 302, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Paulsson, J.F.; Schultz, S.W.; Kohler, M.; Leibiger, I.; Berggren, P.O.; Westermark, G.T. Real-time monitoring of apoptosis by caspase-3-like protease induced FRET reduction triggered by amyloid aggregation. Exp. Diabetes. Res. 2008, 2008, 865850. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of bid by caspase 8 mediates the mitochondrial damage in the fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Wang, K.; Yin, X.M.; Chao, D.T.; Milliman, C.L.; Korsmeyer, S.J. Bid: A novel bh3 domain-only death agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and datp-dependent formation of apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Kominami, K.; Nakabayashi, J.; Nagai, T.; Tsujimura, Y.; Chiba, K.; Kimura, H.; Miyawaki, A.; Sawasaki, T.; Yokota, H.; Manabe, N.; et al. The molecular mechanism of apoptosis upon caspase-8 activation: Quantitative experimental validation of a mathematical model. Biochim. Biophys. Acta 2012, 1823, 1825–1840. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the rip1-rip3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. Rip3, an energy metabolism regulator that switches tnf-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein hmgb1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Li, L.; Qiu, S.; Lu, Q.; Pan, Q.; Gu, Y.; Luo, J.; Hu, X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol. Cancer Ther. 2007, 6, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; White, E. Role of autophagy in cancer: Management of metabolic stress. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kominami, E.; Tanaka, K. Autophagy and neurodegeneration. Autophagy 2006, 2, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.G.; Honson, N.S.; Arns, S.; Davis, T.L.; Dhe-Paganon, S.; Kovacic, S.; Kumar, N.S.; Pfeifer, T.A.; Young, R.N. Development of fluorescent substrates and assays for the key autophagy-related cysteine protease enzyme, ATG4B. Assay Drug Dev. Technol. 2014, 12, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.D.; Matrisian, L.M. The other side of MMPS: Protective roles in tumor progression. Cancer Metastasis Rev. 2007, 26, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Palavalli, L.H.; Prickett, T.D.; Wunderlich, J.R.; Wei, X.; Burrell, A.S.; Porter-Gill, P.; Davis, S.; Wang, C.; Cronin, J.C.; Agrawal, N.S.; et al. Analysis of the matrix metalloproteinase family reveals that mmp8 is often mutated in melanoma. Nat. Genet. 2009, 41, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Fernandez, A.; Fueyo, A.; Folgueras, A.R.; Garabaya, C.; Pennington, C.J.; Pilgrim, S.; Edwards, D.R.; Holliday, D.L.; Jones, J.L.; Span, P.N.; et al. Matrix metalloproteinase-8 functions as a metastasis suppressor through modulation of tumor cell adhesion and invasion. Cancer Res. 2008, 68, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Seiki, M. MT1-MMP: A potent modifier of pericellular microenvironment. J. Cell Physiol 2006, 206, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, Y.; Huang, H.; Pan, Y.; Chaney, E.J.; Boppart, S.A.; Ozer, H.; Strongin, A.Y. Quantitative FRET imaging to visualize the invasiveness of live breast cancer cells. PLoS ONE 2013, 8, e58569. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Lin, J.; Lu, J.; Liu, B.F.; Zeng, S.; Luo, Q. Detection of mmp activity in living cells by a genetically encoded surface-displayed FRET sensor. Biochim. Biophys. Acta 2007, 1773, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Vandame, P.; Spriet, C.; Riquet, F.; Trinel, D.; Cailliau-Maggio, K.; Bodart, J.F. Optimization of ERK activity biosensors for both ratiometric and lifetime FRET measurements. Sensors 2013, 13, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Vandame, P.; Spriet, C.; Trinel, D.; Gelaude, A.; Caillau, K.; Bompard, C.; Biondi, E.; Bodart, J.F. The spatio-temporal dynamics of PKA activity profile during mitosis and its correlation to chromosome segregation. Cell Cycle 2014, 13, 3232–3240. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Miyawaki, A. A high-throughput method for development of FRET-based indicators for proteolysis. Biochem. Biophys. Res. Commun. 2004, 319, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.D.; Gao, L.; Ustione, A.; Bedard, N.; Kester, R.; Piston, D.W.; Tkaczyk, T.S. Real-time hyperspectral fluorescence imaging of pancreatic beta-cell dynamics with the image mapping spectrometer. J. Cell Sci. 2012, 125, 4833–4840. [Google Scholar] [CrossRef] [PubMed]
- Depry, C.; Zhang, J. Visualization of kinase activity with FRET-based activity biosensors. Curr. Protoc. Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Harvey, C.D.; Ehrhardt, A.G.; Cellurale, C.; Zhong, H.; Yasuda, R.; Davis, R.J.; Svoboda, K. A genetically encoded fluorescent sensor of ERK activity. Proc. Natl. Acad. Sci. USA 2008, 105, 19264–19269. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Kumagai, Y.; Sakurai, A.; Komatsu, N.; Fujita, Y.; Shionyu, C.; Matsuda, M. Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 2013, 52, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiratsuka, T.; Fujita, Y.; Naoki, H.; Aoki, K.; Kamioka, Y.; Matsuda, M. Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. Elife 2015, 4, e05178. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.Y.; Kain, K.H.; Klemke, R.L.; Tsien, R.Y. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc. Natl. Acad. Sci. USA 2001, 98, 15003–15008. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Zhang, J. Dynamic visualization of calcium-dependent signaling in cellular microdomains. Cell Calcium 2015, 58, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Griesbeck, O.; Heim, R.; Tsien, R.Y. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc. Natl. Acad. Sci. USA 1999, 96, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Truong, K.; Sawano, A.; Mizuno, H.; Hama, H.; Tong, K.I.; Mal, T.K.; Miyawaki, A.; Ikura, M. FRET-based in vivo Ca2+ imaging by a new calmodulin-gfp fusion molecule. Nat. Struct. Biol. 2001, 8, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; de Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, BCR, on chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Feller, S.M. CRK family adaptors-signalling complex formation and biological roles. Oncogene 2001, 20, 6348–6371. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Frame, M.C. SRC in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Morton, J.P.; Karim, S.A.; Graham, K.; Timpson, P.; Jamieson, N.; Athineos, D.; Doyle, B.; McKay, C.; Heung, M.Y.; Oien, K.A.; et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2010, 139, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, D.P.; Schwartzberg, P.L.; Venegas, A.; Tse, R.; Sheetz, M.P. Selective regulation of integrin—cytoskeleton interactions by the tyrosine kinase src. Nat. Cell Biol. 1999, 1, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Critchley, D.R. Focal adhesions - the cytoskeletal connection. Curr. Opin. Cell Biol. 2000, 12, 133–139. [Google Scholar] [CrossRef]
- Horejsi, V.; Zhang, W.; Schraven, B. Transmembrane adaptor proteins: Organizers of immunoreceptor signalling. Nat. Rev. Immunol. 2004, 4, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, G.; Germain, R.N. Modeling t cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005, 3, e356. [Google Scholar] [CrossRef] [PubMed]
- Cadra, S.; Gucciardi, A.; Valignat, M.P.; Theodoly, O.; Vacaflores, A.; Houtman, J.C.; Lellouch, A.C. Roza-xl, an improved FRET based biosensor with an increased dynamic range for visualizing zeta associated protein 70 kd (zap-70) tyrosine kinase activity in live t cells. Biochem. Biophys. Res. Commun. 2015, 459, 405–410. [Google Scholar] [CrossRef] [PubMed]
- van Beek, J.D.; Hess, S.; Vollrath, F.; Meier, B.H. The molecular structure of spider dragline silk: Folding and orientation of the protein backbone. Proc. Natl. Acad. Sci. USA 2002, 99, 10266–10271. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Helmke, B.P.; Blackman, B.R.; Schwartz, M.A. Mechanisms of mechanotransduction. Dev. Cell 2006, 10, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Alonso, J.L.; Ostuni, E.; Whitesides, G.M.; Ingber, D.E. Cell shape provides global control of focal adhesion assembly. Biochem. Biophys. Res. Commun. 2003, 307, 355–361. [Google Scholar] [CrossRef]
- Bershadsky, A.D.; Balaban, N.Q.; Geiger, B. Adhesion-dependent cell mechanosensitivity. Annu. Rev. Cell. Dev. Biol. 2003, 19, 677–695. [Google Scholar] [CrossRef] [PubMed]
- Becker, N.; Oroudjev, E.; Mutz, S.; Cleveland, J.P.; Hansma, P.K.; Hayashi, C.Y.; Makarov, D.E.; Hansma, H.G. Molecular nanosprings in spider capture-silk threads. Nat. Mater. 2003, 2, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell. Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Cost, A.L.; Ringer, P.; Chrostek-Grashoff, A.; Grashoff, C. How to measure molecular forces in cells: A guide to evaluating genetically-encoded FRET-based tension sensors. Cell. Mol. Bioeng. 2015, 8, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Drepper, T.; Huber, R.; Heck, A.; Circolone, F.; Hillmer, A.K.; Buchs, J.; Jaeger, K.E. Flavin mononucleotide-based fluorescent reporter proteins outperform green fluorescent protein-like proteins as quantitative in vivo real-time reporters. Appl. Environ. Microbiol. 2010, 76, 5990–5994. [Google Scholar] [CrossRef] [PubMed]
- White, A. Effect of PH on fluorescence of tryosine, tryptophan and related compounds. Biochem. J. 1959, 71, 217–220. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hochreiter, B.; Pardo-Garcia, A.; Schmid, J.A. Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences. Sensors 2015, 15, 26281-26314. https://doi.org/10.3390/s151026281
Hochreiter B, Pardo-Garcia A, Schmid JA. Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences. Sensors. 2015; 15(10):26281-26314. https://doi.org/10.3390/s151026281
Chicago/Turabian StyleHochreiter, Bernhard, Alan Pardo-Garcia, and Johannes A. Schmid. 2015. "Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences" Sensors 15, no. 10: 26281-26314. https://doi.org/10.3390/s151026281
APA StyleHochreiter, B., Pardo-Garcia, A., & Schmid, J. A. (2015). Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences. Sensors, 15(10), 26281-26314. https://doi.org/10.3390/s151026281