
Characterization of Biosensors Based on Recombinant Glutamate Oxidase: Comparison of Crosslinking Agents in Terms of Enzyme Loading and Efficiency Parameters


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Instrumentation and Software
2.3. Amperometric Experiments
2.4. Electrode Preparation and Modification
2.4.1. Electrode Preparation
2.4.2. Electrode Modification
2.5. Biosensor Nomenclature
2.6. Data Analysis
2.6.1. Hydrogen Peroxide Sensitivity Ratio, HP%
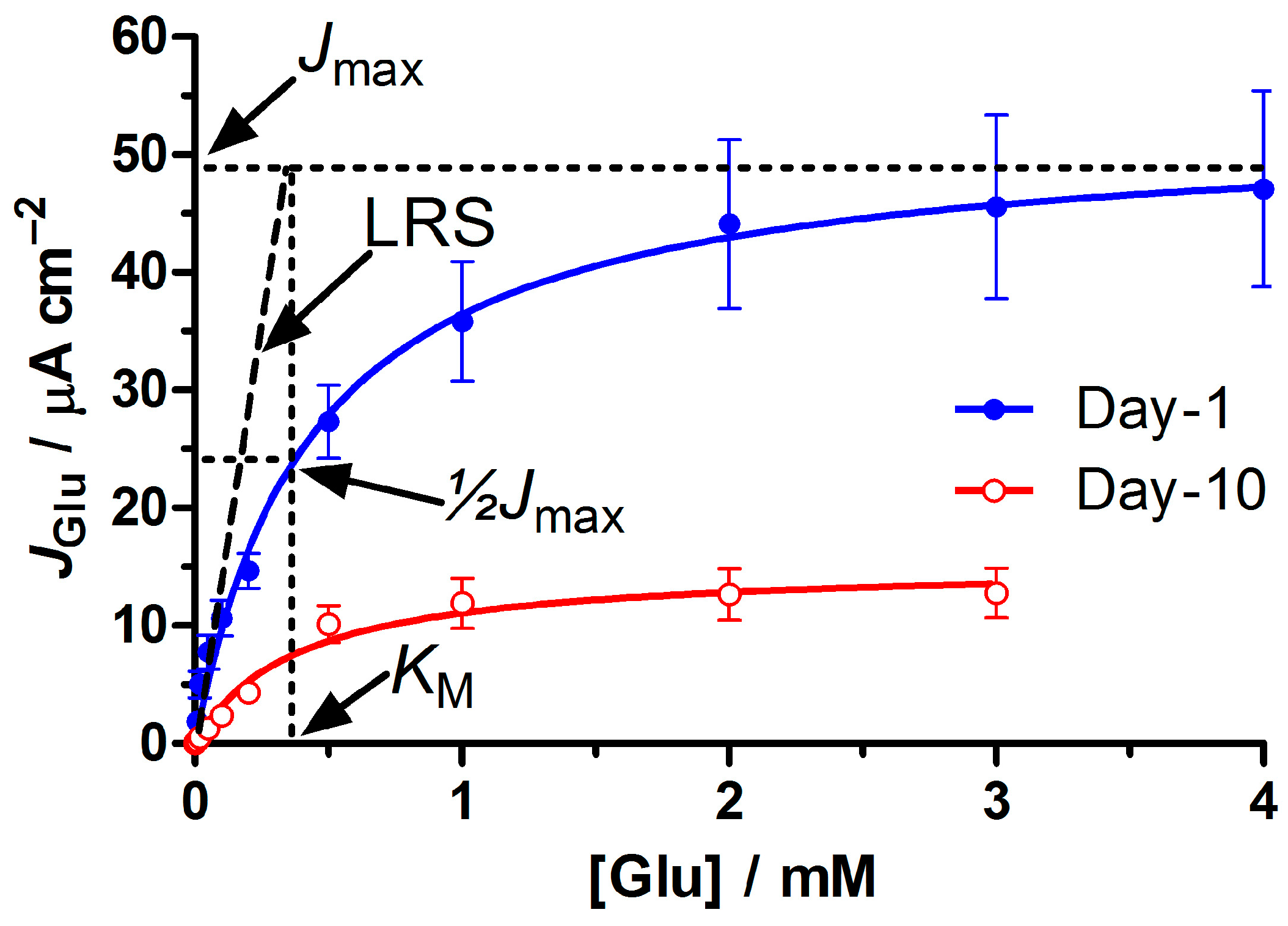
2.6.2. Michaelis-Menten Parameters
2.6.3. Normalized Enzyme Parameters, BE% and Enzact
2.7. Statistical Analysis
3. Results and Discussion
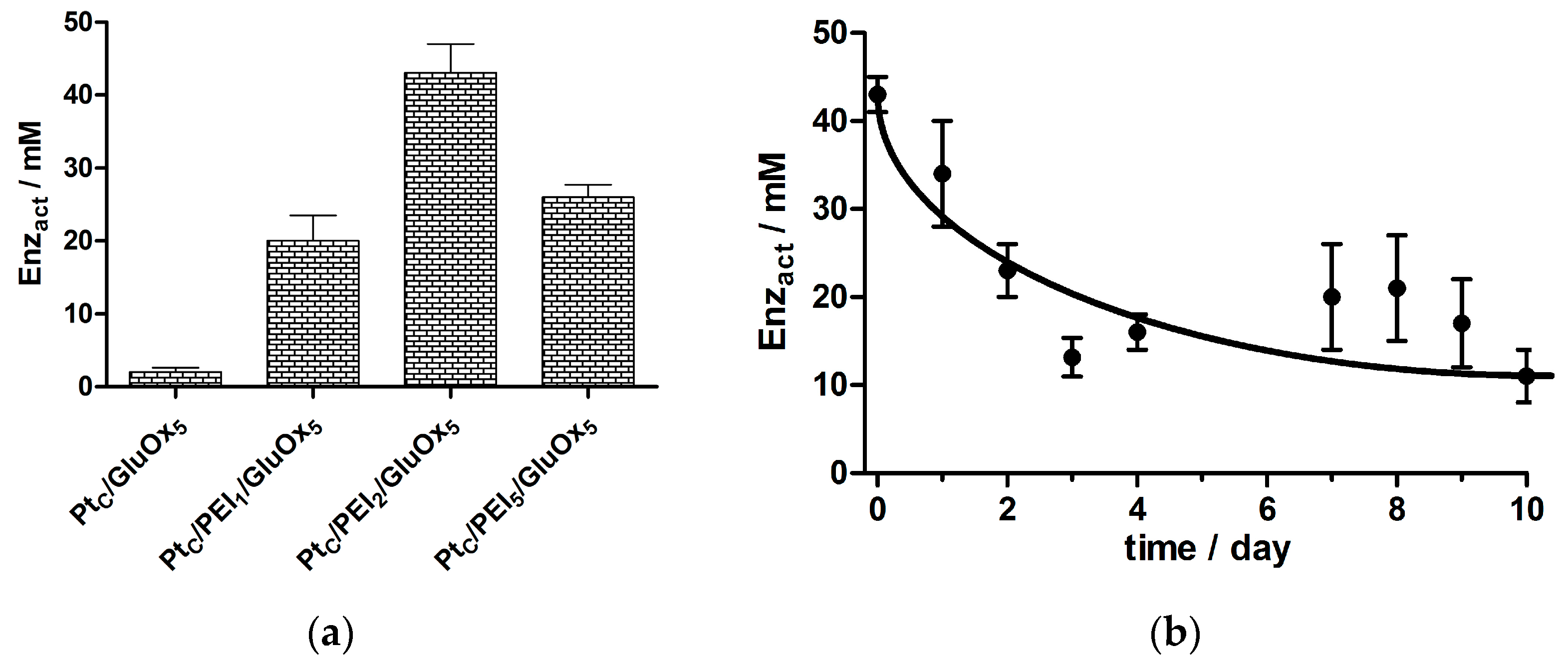
3.1. Optimizing the Ratio of PEI to GluOx
3.1.1. GluOx Loading Parameter, Enzact
3.1.2. Biosensor Efficiency Parameter, BE%
3.2. Biosensor Performance in the Presence and Absence of the Permselective PoPD Layer
3.3. Within-Study Comparison of Native and Recombinant GluOx Forms
3.4. Glutaraldehyde
Effect of Glutaraldehyde on BE%
3.5. Effect of Other Crosslinking Agents on BE%
3.5.1. Polyethylene Glycol, PEG
3.5.2. Poly(ethyleneglycol) Diglycidyl Ether, PEGDE
3.6. Limit of Detection, and Response Time
3.7. Stability of Selected Biosensor Designs up to Day 90
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prodromidis, M.I.; Karayannis, M.I. Enzyme based amperometric biosensors for food analysis. Electroanalysis 2002, 14, 241–261. [Google Scholar] [CrossRef]
- Bahadir, E.B.; Sezginturk, M.K. Applications of commercial biosensors in clinical, food, environmental, and biothreat/biowarfare analyses. Anal. Biochem. 2015, 478, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Mello, L.D.; Kubota, L.T. Review of the use of biosensors as analytical tools in the food and drink industries. Food Chem. 2002, 77, 237–256. [Google Scholar] [CrossRef]
- Kennedy, R.T. Emerging trends in in vivo neurochemical monitoring by microdialysis. Curr. Opin. Chem. Biol. 2013, 17, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hascup, K.N.; Hascup, E.R. Electrochemical techniques for subsecond neurotransmitter detection in live rodents. Comp. Med. 2014, 64, 249–255. [Google Scholar] [PubMed]
- Marsman, A.; van den Heuvel, M.P.; Klomp, D.W.J.; Kahn, R.S.; Luijten, P.R.; Pol, H.E.H. Glutamate in schizophrenia: A focused review and meta-analysis of H-1-MRS studies. Schizophr. Bull. 2013, 39, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.W.; Kornhuber, J.; Riederer, P. Dopamine/glutamate interactions in Parkinson’s disease. Neurosci. Biobehav. Rev. 1997, 21, 393–400. [Google Scholar] [CrossRef]
- Davalos, A.; Castillo, J.; Serena, J.; Noya, M. Duration of glutamate release after acute ischemic stroke. Stroke 1997, 28, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.G.; Brandt, L.; Ungerstedt, U.; Saveland, H. Bedside detection of brain ischemia using intracerebral microdialysis: Subarachnoid hemorrhage and delayed ischemic deterioration. Neurosurgery 1999, 45, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Saveland, H.; Nilsson, O.G.; BorisMoller, F.; Wieloch, T.; Brandt, L. Intracerebral microdialysis of glutamate and aspartate in two vascular territories after aneurysmal subarachnoid hemorrhage. Neurosurgery 1996, 38, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.; Berners, M.; Boutelle, M.G.; Kusakabe, H.; Fillenz, M. The determination of the extracellular concentration of brain glutamate using quantitative microdialysis. Brain Res. 1996, 707, 131–133. [Google Scholar] [CrossRef]
- Shaw, P.J.; Forrest, V.; Ince, P.G.; Richardson, J.P.; Wastell, H.J. CSF and plasma amino-acid levels in motor-neuron disease—Elevation of CSF glutamate in a subset of patients. Neurodegeneration 1995, 4, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.T.; Thompson, J.E.; Vickroy, T.W. In vivo monitoring of amino acids by direct sampling of brain extracellular fluid at ultralow flow rates and capillary electrophoresis. J. Neurosci. Methods 2002, 114, 39–49. [Google Scholar] [CrossRef]
- Herman, M.A.; Jahr, C.E. Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 2007, 27, 9736–9741. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Swedlow, P.E.; Styren, S.D.; DeKosky, S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993, 61, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.W.; Bargmann, C.I.; et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Fillenz, M. In vivo neurochemical monitoring and the study of behaviour. Neurosci. Biobehav. Rev. 2005, 29, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Van der Zeyden, M.; Denziel, W.H.; Rea, K.; Cremers, T.I.; Westerink, B.H. Microdialysis of GABA and glutamate: Analysis, interpretation and comparison with microsensors. Pharmacol. Biochem. Behav. 2008, 90, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Rocchitta, G.; Spanu, A.; Babudieri, S.; Latte, G.; Madeddu, G.; Galleri, G.; Nuvoli, S.; Bagella, P.; Demartis, M.I.; Fiore, V.; et al. Enzyme biosensors for biomedical applications: Strategies for safeguarding analytical performances in biological fluids. Sensors 2016, 16, 780–800. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, R.D.; Lowry, J.P.; Rocchitta, G.; McMahon, C.P.; Serra, P.A. Designing sensitive and selective polymer/enzyme composite biosensors for brain monitoring in vivo. Trends Anal. Chem. 2008, 27, 78–88. [Google Scholar] [CrossRef]
- Zhang, J.; Jaquins-Gerstl, A.; Nesbitt, K.M.; Rutan, S.C.; Michael, A.C.; Weber, S.G. In vivo monitoring of serotonin in the striatum of freely moving rats with one minute temporal resolution by online microdialysis-capillary high-performance liquid chromatography at elevated temperature and pressure. Anal. Chem. 2013, 85, 9889–9897. [Google Scholar] [CrossRef] [PubMed]
- Crick, E.W.; Osorio, I.; Frei, M.; Mayer, A.P.; Lunte, C.E. Correlation of 3-mercaptopropionic acid induced seizures and changes in striatal neurotransmitters monitored by microdialysis. Eur. J. Pharm. Sci. 2014, 57, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.J.; Sandberg, S.G.; Wanat, M.J.; Gan, J.O.; Horne, E.A.; Hart, A.S.; Akers, C.A.; Parker, J.G.; Willuhn, I.; Martinez, V.; et al. Chronic microsensors for longitudinal, subsecond dopamine detection in behaving animals. Nat. Methods 2010, 7, 126–129. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, R.D. Long-term monitoring of brain dopamine metabolism in vivo with carbon paste electrodes. Sensors 2005, 5, 317–342. [Google Scholar] [CrossRef]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Immobilization strategies to develop enzymatic biosensors. Biotechnol. Adv. 2012, 30, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, S.; McNeil, C.J.; Lowry, J.P.; McMahon, C.P.; O’Neill, R.D. Highly selective and stable microdisc biosensors for l-glutamate monitoring. Sens. Actuator B Chem. 2013, 178, 606–614. [Google Scholar] [CrossRef]
- Salazar, P.; Martin, M.; O’Neill, R.D.; Roche, R.; Gonzalez-Mora, J.L. Improvement and characterization of surfactant-modified Prussian Blue screen-printed carbon electrodes for selective H2O2 detection at low applied potentials. J. Electroanal. Chem. 2012, 674, 48–56. [Google Scholar] [CrossRef]
- Hamdi, N.; Wang, J.J.; Walker, E.; Maidment, N.T.; Monbouquette, H.G. An electroenzymatic l-glutamate microbiosensor selective against dopamine. J. Electroanal. Chem. 2006, 591, 33–40. [Google Scholar] [CrossRef]
- White, S.F.; Turner, A.P.F.; Schmid, R.D.; Bilitewski, U.; Bradley, J. Investigations of platinized and rhodinized carbon electrodes for use in glucose sensors. Electroanalysis 1994, 6, 625–632. [Google Scholar] [CrossRef]
- Sasso, S.V.; Pierce, R.J.; Walla, R.; Yacynych, A.M. Electropolymerized 1,2-diaminobenzene as a means to prevent interferences and fouling and to stabilize immobilized enzyme in electrochemical biosensors. Anal. Chem. 1990, 62, 1111–1117. [Google Scholar] [CrossRef]
- Lowry, J.P.; O'Neill, R.D. Partial characterization in vitro of glucose oxidase-modified poly(phenylenediamine)-coated electrodes for neurochemical analysis in vivo. Electroanalysis 1994, 6, 369–379. [Google Scholar] [CrossRef]
- Salazar, P.; Martin, M.; Roche, R.; Gonzalez-Mora, J.L.; O’Neill, R.D. Microbiosensors for glucose based on Prussian Blue modified carbon fiber electrodes for in vivo monitoring in the central nervous system. Biosens. Bioelectron. 2010, 26, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Wahono, N.; Qin, S.; Oomen, P.; Cremers, T.I.F.; de Vries, M.G.; Westerink, B.H.C. Evaluation of permselective membranes for optimization of intracerebral amperometric glutamate biosensors. Biosens. Bioelectron. 2012, 33, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Rocchitta, G.; Secchi, O.; Alvau, M.D.; Migheli, R.; Calia, G.; Bazzu, G.; Farina, D.; Desole, M.S.; O'Neill, R.D.; Serra, P.A. Development and characterization of an implantable biosensor for telemetric monitoring of ethanol in the brain of freely moving rats. Anal. Chem. 2012, 84, 7072–7079. [Google Scholar] [CrossRef] [PubMed]
- Secchi, O.; Zinellu, M.; Spissu, Y.; Pirisinu, M.; Bazzu, G.; Migheli, R.; Desole, M.S.; O'Neill, R.D.; Serra, P.A.; Rocchitta, G. Further in vitro characterization of an implantable biosensor for ethanol monitoring in the brain. Sensors 2013, 13, 9522–9535. [Google Scholar] [CrossRef] [PubMed]
- Arrigan, D.W.M.; Bartlett, P.N. A scanning force microscopy study of poly(phenol) films containing immobilized glucose oxidase. Biosens. Bioelectron. 1998, 13, 293–304. [Google Scholar] [CrossRef]
- Cosnier, S. Biomolecule immobilization on electrode surfaces by entrapment or attachment to electrochemically polymerized films. A review. Biosens. Biolelectron. 1999, 14, 443–456. [Google Scholar] [CrossRef]
- Craig, J.D.; O'Neill, R.D. Electrosynthesis and permselective characterisation of phenol-based polymers for biosensor applications. Anal. Chim. Acta 2003, 495, 33–43. [Google Scholar] [CrossRef]
- Calia, G.; Monti, P.; Marceddu, S.; Dettori, M.A.; Fabbri, D.; Jaoua, S.; O’Neill, R.D.; Serra, P.A.; Delogu, G.; Migheli, Q. Electropolymerized phenol derivatives as permselective polymers for biosensor applications. Analyst 2015, 140, 3607–3615. [Google Scholar] [CrossRef] [PubMed]
- Fillenz, M.; O'Neill, R.D. Effects of light reversal on the circadian pattern of motor activity and voltammetric signals recorded in rat forebrain. J. Physiol. 1986, 374, 91–101. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, R.D.; Fillenz, M.; Albery, W.J. Circadian changes in homovanillic acid and ascorbate levels in the rat striatum using microprocessor-controlled voltammetry. Neurosci. Lett. 1982, 34, 189–193. [Google Scholar] [CrossRef]
- Murphy, L.J. Reduction of interference response at a hydrogen peroxide detecting electrode using electropolymerized films of substituted naphthalenes. Anal. Chem. 1998, 70, 2928–2935. [Google Scholar] [CrossRef]
- Dai, Y.Q.; Zhou, D.M.; Shiu, K.K. Permeability and permselectivity of polyphenylenediamine films synthesized at a palladium disk electrode. Electrochim. Acta 2006, 52, 297–303. [Google Scholar] [CrossRef]
- Kirwan, S.M.; Rocchitta, G.; McMahon, C.P.; Craig, J.D.; Killoran, S.J.; O'Brien, K.B.; Serra, P.A.; Lowry, J.P.; O'Neill, R.D. Modifications of poly(O-phenylenediamine) permselective layer on Pt-Ir for biosensor application in neurochemical monitoring. Sensors 2007, 7, 420–437. [Google Scholar] [CrossRef]
- Rothwell, S.A.; O'Neill, R.D. Effects of applied potential on the mass of non-conducting poly(ortho-phenylenediamine) electro-deposited on EQCM electrodes: Comparison with biosensor selectivity parameters. Phys. Chem. Chem. Phys. 2011, 13, 5413–5421. [Google Scholar] [CrossRef] [PubMed]
- Killoran, S.J.; O'Neill, R.D. Characterization of permselective coatings electrosynthesized on Pt-Ir from the three phenylenediamine isomers for biosensor applications. Electrochim. Acta 2008, 53, 7303–7312. [Google Scholar] [CrossRef]
- McMahon, C.P.; Rocchitta, G.; Serra, P.A.; Kirwan, S.M.; Lowry, J.P.; O'Neill, R.D. The efficiency of immobilised glutamate oxidase decreases with surface enzyme loading: an electrostatic effect, and reversal by a polycation significantly enhances biosensor sensitivity. Analyst 2006, 131, 68–72. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.P.; Rocchitta, G.; Kirwan, S.M.; Killoran, S.J.; Serra, P.A.; Lowry, J.P.; O'Neill, R.D. Oxygen tolerance of an implantable polymer/enzyme composite glutamate biosensor displaying polycation-enhanced substrate sensitivity. Biosens. Bioelectron. 2007, 22, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, S.A.; Kinsella, M.E.; Zain, Z.M.; Serra, P.A.; Rocchitta, G.; Lowry, J.P.; O'Neill, R.D. Contributions by a novel edge effect to the permselectivity of an electrosynthesized polymer for microbiosensor applications. Anal. Chem. 2009, 81, 3911–3918. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.P.; Rocchitta, G.; Serra, P.A.; Kirwan, S.M.; Lowry, J.P.; O’Neill, R.D. Control of the oxygen dependence of an implantable polymer/enzyme composite biosensor for glutamate. Anal. Chem. 2006, 78, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.R.; Lowry, J.P.; Oneill, R.D. Biosensor for neurotransmitter l-glutamic acid designed for efficient use of l-glutamate oxidase and effective rejection of interference. Analyst 1997, 122, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Lowry, J.P.; Ryan, M.R.; O’Neill, R.D. Behaviourally induced changes in extracellular levels of brain glutamate monitored at 1 s resolution with an implanted biosensor. Anal. Commun. 1998, 35, 87–89. [Google Scholar] [CrossRef]
- Arima, J.; Tamura, T.; Kusakabe, H.; Ashiuchi, M.; Yagi, T.; Tanaka, H.; Inagaki, K. Recombinant expression, biochemical characterization and stabilization through proteolysis of an l-glutamate oxidase from Streptomyces sp X-119-6. J. Biochem. 2003, 134, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, H.; Midorikawa, Y.; Fujishima, T.; Kuninaka, A.; Yoshino, H. Purification and properties of a new enzyme, l-glutamate oxidase, from Streptomyces sp. X-119-6 grown on wheat bran. Agric. Biol. Chem. 1983, 47, 1323–1328. [Google Scholar] [CrossRef]
- Mikeladze, E.; Collins, A.; Sukhacheva, M.; Netrusov, A.; Csoregi, E. Characterization of a glutamate biosensor based on a novel glutamate oxidase integrated into a redox hydrogel. Electroanalysis 2002, 14, 1052–1059. [Google Scholar] [CrossRef]
- Sirca, D.; Vardeu, A.; Pinna, M.; Diana, M.; Enrico, P. A robust, state-of-the-art amperometric microbiosensor for glutamate detection. Biosens. Bioelectron. 2014, 61, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Vasylieva, N.; Maucler, C.; Meiller, A.; Viscogliosi, H.; Lieutaud, T.; Barbier, D.; Marinesco, S. Immobilization method to preserve enzyme specificity in biosensors: Consequences for brain glutamate detection. Anal. Chem. 2013, 85, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.T.; Chang, C.F.; Chan, W.C. Fabrication of implantable, enzyme-immobilized glutamate sensors for the monitoring of glutamate concentration changes in vitro and in vivo. Molecules 2014, 19, 7341–7355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mullens, C.; Gorski, W. Amperometric glutamate biosensor based on chitosan enzyme film. Electrochim. Acta 2006, 51, 4528–4532. [Google Scholar] [CrossRef]
- Khan, A.S.; Michael, A.C. Invasive consequences of using micro-electrodes and microdialysis probes in the brain. Trends Anal. Chem. 2003, 22, 503–508. [Google Scholar] [CrossRef]
- Jaquins-Gerstl, A.; Shu, Z.; Zhang, J.; Liu, Y.; Weber, S.G.; Michael, A.C. Effect of dexamethasone on gliosis, ischemia, and dopamine extraction during microdialysis sampling in brain tissue. Anal. Chem. 2011, 83, 7662–7667. [Google Scholar] [CrossRef] [PubMed]
- Clapp-Lilly, K.L.; Roberts, R.C.; Duffy, L.K.; Irons, K.P.; Hu, Y.; Drew, K.L. An ultrastructural analysis of tissue surrounding a microdialysis probe. J. Neurosci. Methods 1999, 90, 129–142. [Google Scholar] [CrossRef]
- Hascup, E.R.; af Bjerken, S.; Hascup, K.N.; Pomerleau, F.; Huettl, P.; Stromberg, I.; Gerhardt, G.A. Histological studies of the effects of chronic implantation of ceramic-based microelectrode arrays and microdialysis probes in rat prefrontal cortex. Brain Res. 2009, 1291, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, S.A.; McMahon, C.P.; O'Neill, R.D. Effects of polymerization potential on the permselectivity of poly(o-phenylenediamine) coatings deposited on Pt-Ir electrodes for biosensor applications. Electrochim. Acta 2010, 55, 1051–1060. [Google Scholar] [CrossRef]
- Wei, W.; Song, Y.; Shi, W.; Lin, N.; Jiang, T.; Cai, X. A high sensitivity MEA probe for measuring real time rat brain glucose flux. Biosens. Biolelectron. 2014, 55, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Lowry, J.P.; McAteer, K.; El Atrash, S.S.; Duff, A.; O'Neill, R.D. Characterization of glucose oxidase-modified poly(phenylenediamine)-coated electrodes in vitro and in vivo: Homogeneous interference by ascorbic acid in hydrogen peroxide detection. Anal. Chem. 1994, 66, 1754–1761. [Google Scholar] [CrossRef]
- Vatsyayan, P.; Bordoloi, S.; Goswami, P. Large catalase based bioelectrode for biosensor application. Biophys. Chem. 2010, 153, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.Y.; Yamamoto, K.; Zhou, T.S.; Xu, F.; Kato, T.; Jin, J.Y.; Jin, L.T. On-line biosensors for simultaneous determination of glucose, choline, and glutamate integrated with a microseparation system. Electrophoresis 2003, 24, 3266–3272. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.G.; Erlenkotter, A.; Cammann, K.; Chemnitius, G.C. Fabrication and characterization of disposable type lactate oxidase sensors for dairy products and clinical analysis. Sens. Actuator B Chem. 2000, 67, 134–141. [Google Scholar] [CrossRef]
- Bunik, V.I.; Schloss, J.V.; Pinto, J.T.; Gibson, G.E.; Cooper, A.J.L. Enzyme-catalyzed side reactions with molecular oxygen may contribute to cell signaling and neurodegenerative diseases. Neurochem. Res. 2007, 32, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Reyes De Corcuera, J.I.; Cavalieri, R.P.; Powers, J.R. Improved platinization conditions produce a 60-fold increase in sensitivity of amperometric biosensors using glucose oxidase immobilized in poly-o-phenylenediamine. J. Electroanal. Chem. 2005, 575, 229–241. [Google Scholar] [CrossRef]
- Chirizzi, D.; Malitesta, C. Potentiometric urea biosensor based on urease immobilized by an electrosynthesized poly(o-phenylenediamine) film with buffering capability. Sens. Actuator B Chem. 2011, 157, 211–215. [Google Scholar] [CrossRef]
- Chen, W.; Cai, S.; Ren, Q.Q.; Wen, W.; Zhao, Y.D. Recent advances in electrochemical sensing for hydrogen peroxide: A review. Analyst 2012, 137, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.Y.; Li, M.R.; Xie, Q.J.; Liu, M.L.; Tan, Y.M.; Xu, X.H.; Yao, S.Z. New glucose biosensor based on a poly(o-phenylendiamine)/glucose oxidase-glutaraldehyde/Prussian blue/Au electrode with QCM monitoring of various electrode-surface modifications. Anal. Chim. Acta 2006, 557, 85–94. [Google Scholar] [CrossRef]
- Hamdan, S.K.; Mohd Zain, A. In vivo Electrochemical Biosensor for Brain Glutamate Detection: A Mini Review. Malays. J. Med. Sci. 2014, 21, 12–26. [Google Scholar] [PubMed]
- McMahon, C.P.; O'Neill, R.D. Polymer-enzyme composite biosensor with high glutamate sensitivity and low oxygen dependence. Anal. Chem. 2005, 77, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Kwon, N.H.; Won, M.S.; Choe, E.S.; Shim, Y.S. Functionalized conducting polymer as an enzyme-immobilizing substrate: An amperometric glutamate microbiosensor for in vivo measurements. Anal. Chem. 2005, 77, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Yigzaw, Y.; Gorton, L. Prussian blue-glutamate oxidase modified glassy carbon electrode: A sensitive l-glutamate and β-N-oxalyl-α,β-diaminopropionic acid (β-ODAP) sensor. Anal. Chim. Acta 2006, 556, 319–325. [Google Scholar] [CrossRef]
- Ammam, M.; Fransaer, J. Highly sensitive and selective glutamate microbiosensor based on cast polyurethane/AC-electrophoresis deposited multiwalled carbon nanotubes and then glutamate oxidase/electrosynthesized polypyrrole/Pt electrode. Biosens. Bioelectron. 2010, 25, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Windmiller, J.R.; Valdes-Ramirez, G.; Zhou, N.; Zhou, M.; Miller, P.R.; Jin, C.; Brozik, S.M.; Polsky, R.; Katz, E.; Narayan, R.; Wang, J. Bicomponent microneedle array biosensor for minimally-invasive glutamate monitoring. Electroanalysis 2011, 23, 2302–2309. [Google Scholar] [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [PubMed]
- Turner, A.P.F.; Karube, I.; Wilson, G.S. Biosensors: Fundamentals and Applications; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Zhang, M.G.; Mullens, C.; Gorski, W. Chitosan-glutamate oxidase gels: Synthesis, characterization, and glutamate determination. Electroanalysis 2005, 17, 2114–2120. [Google Scholar] [CrossRef]
- Barsan, M.M.; Klincar, J.; Batic, M.; Brett, C.M.A. Design and application of a flow cell for carbon-film based electrochemical enzyme biosensors. Talanta 2007, 71, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, R.; Chebib, H.; Saikali, Y.; Vittori, O.; Sigaud, M.; Jaffrezic-Renault, N. Amperometric and impedimetric characterization of a glutamate biosensor based on Nafion® and a methyl viologen modified glassy carbon electrode. Biosens. Bioelectron. 2007, 22, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Zalipsky, S. Chemistry of polyethylene-glycol conjugates with biologically-active molecules. Adv. Drug Deliv. Rev. 1995, 16, 157–182. [Google Scholar] [CrossRef]
- Gregg, B.A.; Heller, A. Redox polymer-films containing enzymes. 2. Glucose-oxidase containing enzyme electrodes. J. Phys. Chem. 1991, 95, 5976–5980. [Google Scholar] [CrossRef]
- Vasylieva, N.; Barnych, B.; Meillerd, A.; Maucler, C.; Pollegioni, L.; Lin, J.S.; Barbier, D.; Marinesco, S. Covalent enzyme immobilization by poly(ethylene glycol) diglycidyl ether (PEGDE) for microelectrode biosensor preparation. Biosens. Bioelectron. 2011, 26, 3993–4000. [Google Scholar] [CrossRef] [PubMed]
- Cragg, S.J.; Rice, M.E. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004, 27, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, B.L.; Regehr, W.G. Timing of neurotransmission at fast synapses in the mammalian brain. Nature 1996, 384, 170–172. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Crosslinker | BE% (Day 0) | BE% (Day 10) | ΔBE% (Day 0) | p-Value | 10-Day Stability | p-Value |
|---|---|---|---|---|---|---|
| none | 41 ± 2 (n = 16) | 26 ± 3 (n = 14) | 0 | N/A | −15 ± 4 | <0.001 |
| GA (1%) | 37 ± 1 (n = 2) | 23 ± 2 (n = 2) | −4 ± 2 | >0.61 | −14 ± 3 | <0.03 |
| GA (25%) | 39 ± 3 (n = 8) | 45 ± 3 (n = 6) | −3 ± 4 | >0.57 | +6 ± 4 | >0.19 |
| PEG (0.1%) | 45 ± 2 (n = 3) | 19 ± 6 (n = 3) | +4 ± 3 | >0.41 | −26 ± 5 | <0.02 |
| PEG (1%) | 45 ± 8 (n = 3) | 21 ± 6 (n = 3) | +4 ± 7 | >0.46 | −24 ± 9 | <0.07 |
| PEGDE (0.1%) | 39 ± 2 (n = 7) | 34 ± 3 (n = 7) | −2 ± 3 | >0.55 | −5 ± 4 | >0.28 |
| PEGDE (1%) | 34 ± 2 (n = 12) | 35 ± 3 (n = 12) | −7 ± 3 | <0.05 | +1 ± 4 | >0.78 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ford, R.; Quinn, S.J.; O’Neill, R.D. Characterization of Biosensors Based on Recombinant Glutamate Oxidase: Comparison of Crosslinking Agents in Terms of Enzyme Loading and Efficiency Parameters. Sensors 2016, 16, 1565. https://doi.org/10.3390/s16101565
Ford R, Quinn SJ, O’Neill RD. Characterization of Biosensors Based on Recombinant Glutamate Oxidase: Comparison of Crosslinking Agents in Terms of Enzyme Loading and Efficiency Parameters. Sensors. 2016; 16(10):1565. https://doi.org/10.3390/s16101565
Chicago/Turabian StyleFord, Rochelle, Susan J. Quinn, and Robert D. O’Neill. 2016. "Characterization of Biosensors Based on Recombinant Glutamate Oxidase: Comparison of Crosslinking Agents in Terms of Enzyme Loading and Efficiency Parameters" Sensors 16, no. 10: 1565. https://doi.org/10.3390/s16101565
APA StyleFord, R., Quinn, S. J., & O’Neill, R. D. (2016). Characterization of Biosensors Based on Recombinant Glutamate Oxidase: Comparison of Crosslinking Agents in Terms of Enzyme Loading and Efficiency Parameters. Sensors, 16(10), 1565. https://doi.org/10.3390/s16101565