
A Disposable Microfluidic Device with a Screen Printed Electrode for Mimicking Phase II Metabolism

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemical Reagents
2.2. Instrumentation
2.3. Screen-Printed Electrodes (SPEs)
2.4. Microfluidic Device with an SPE
2.4.1. Time of Electrolysis in the SPE Cell
2.4.2. Time of GSH Conjugation in a Serpentine Channel
2.4.3. Fabrication of Chip
2.5. Optimization of SPEs
2.6. Chip/ESI-MS
3. Validation of SPEs
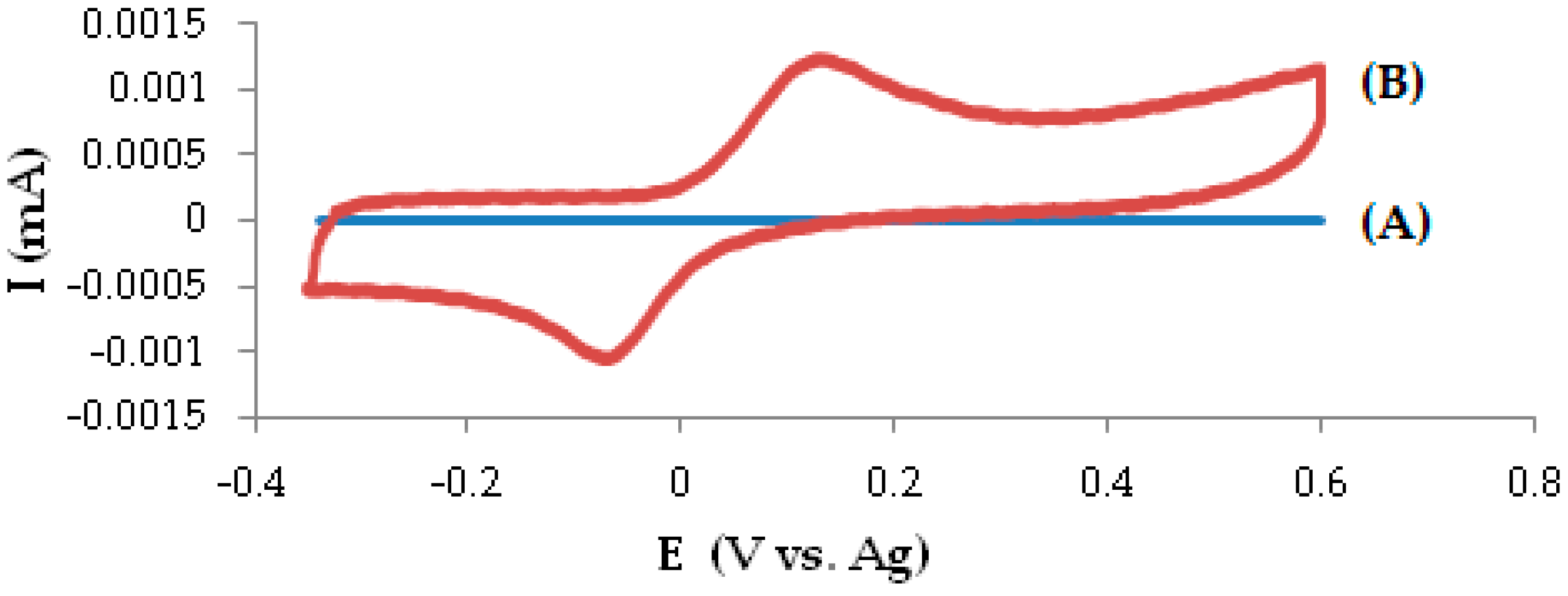
3.1. Electrochemical Behavior of DOPA
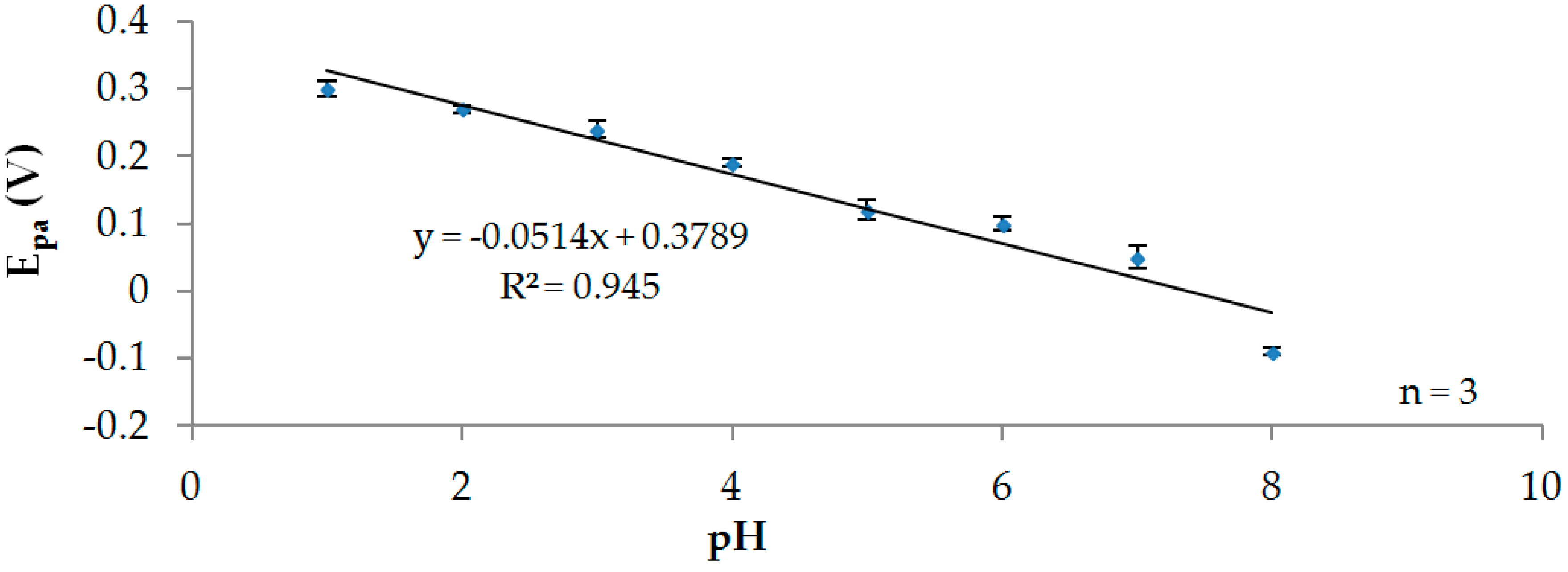
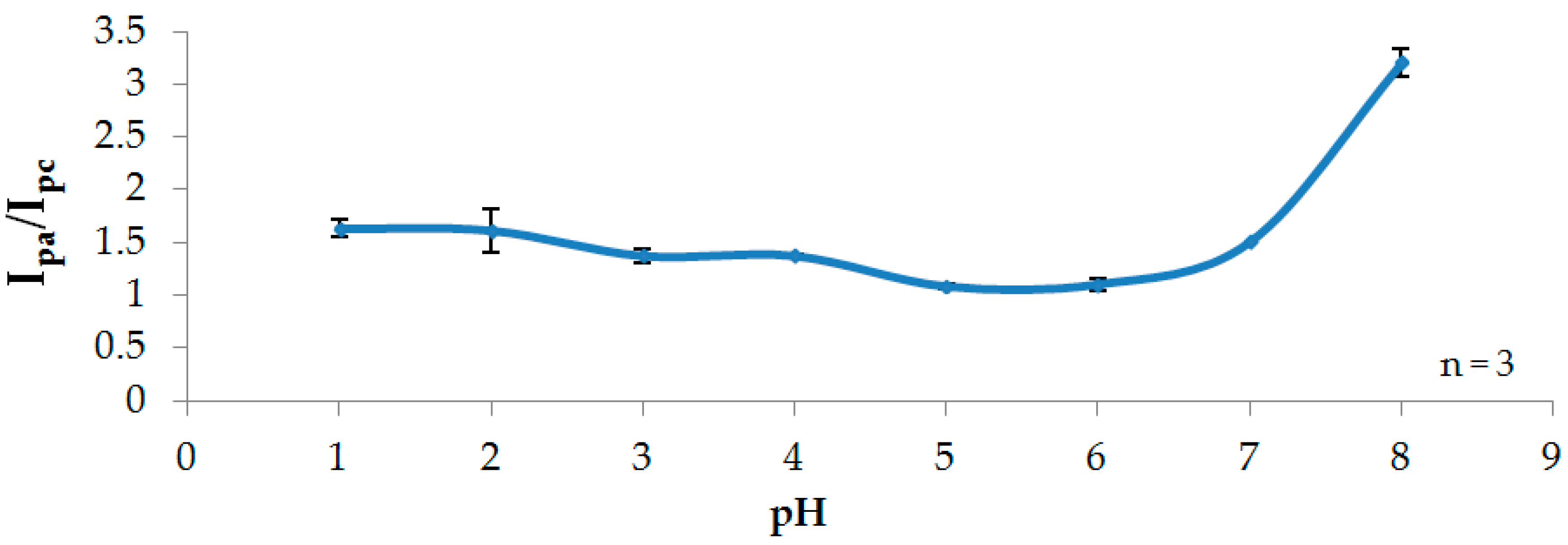
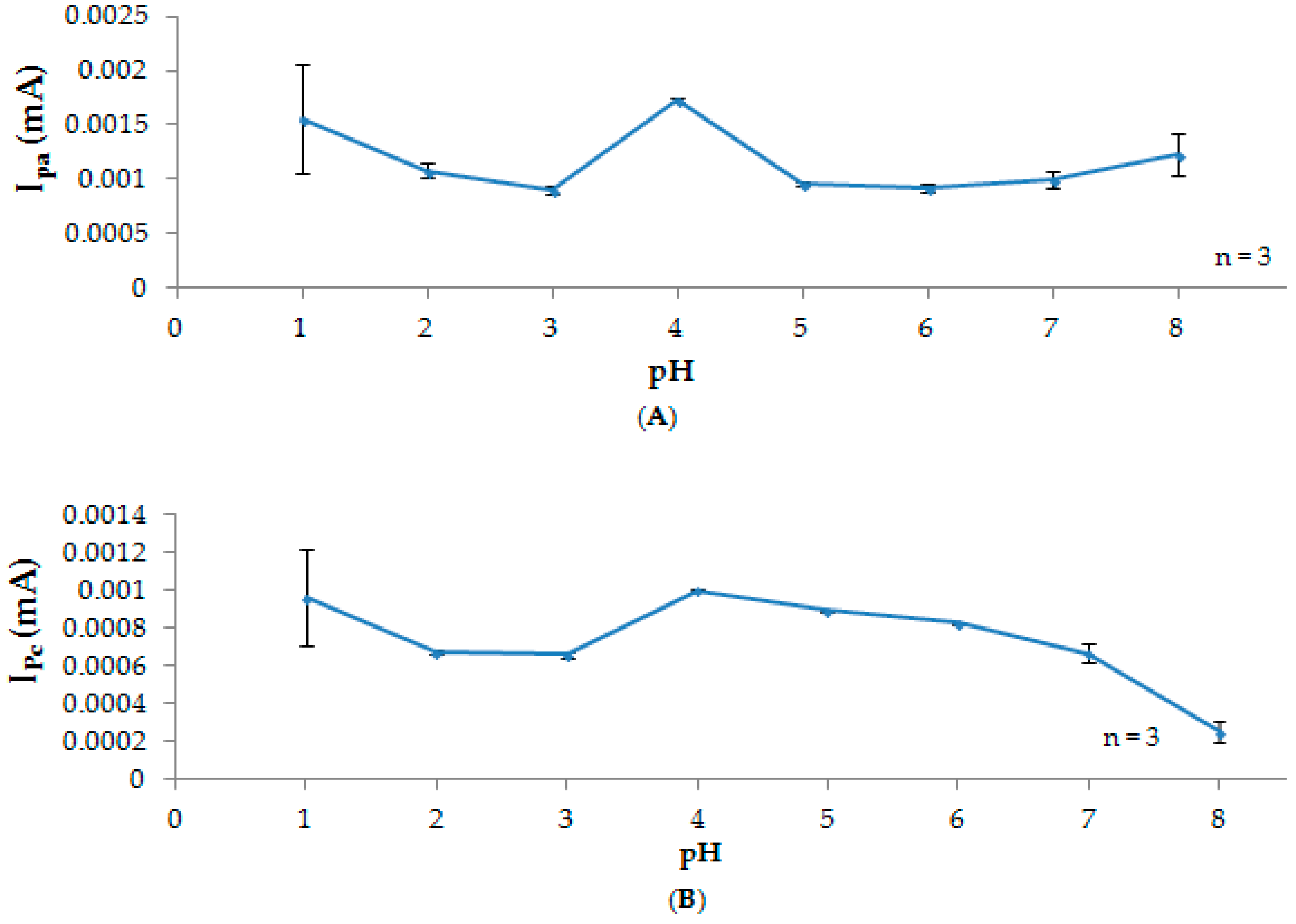
3.2. Effect of pH
3.2.1. Participation of Protons
3.2.2. Stability Studies
3.2.3. Current
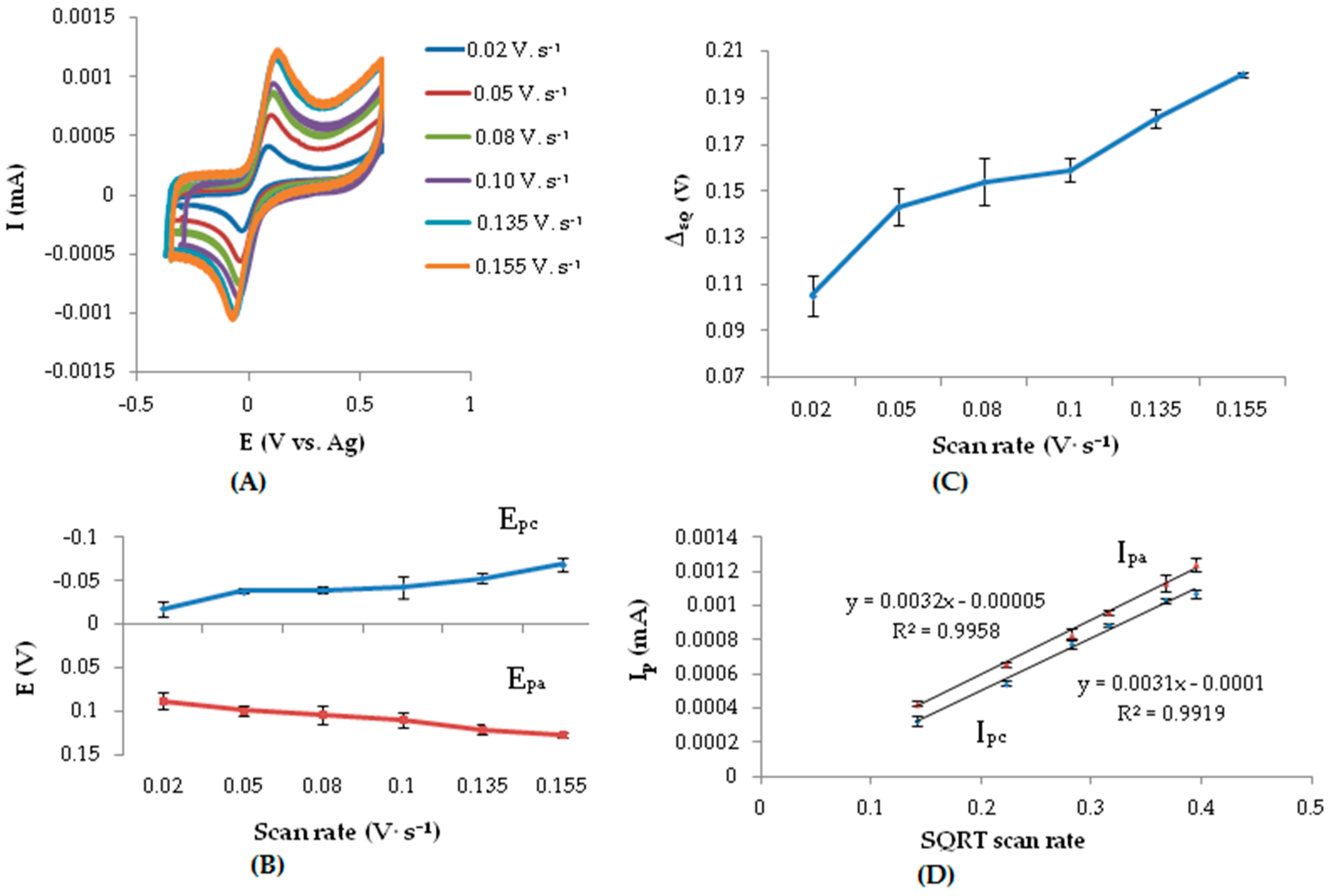
3.3. Effect of Scan Rate
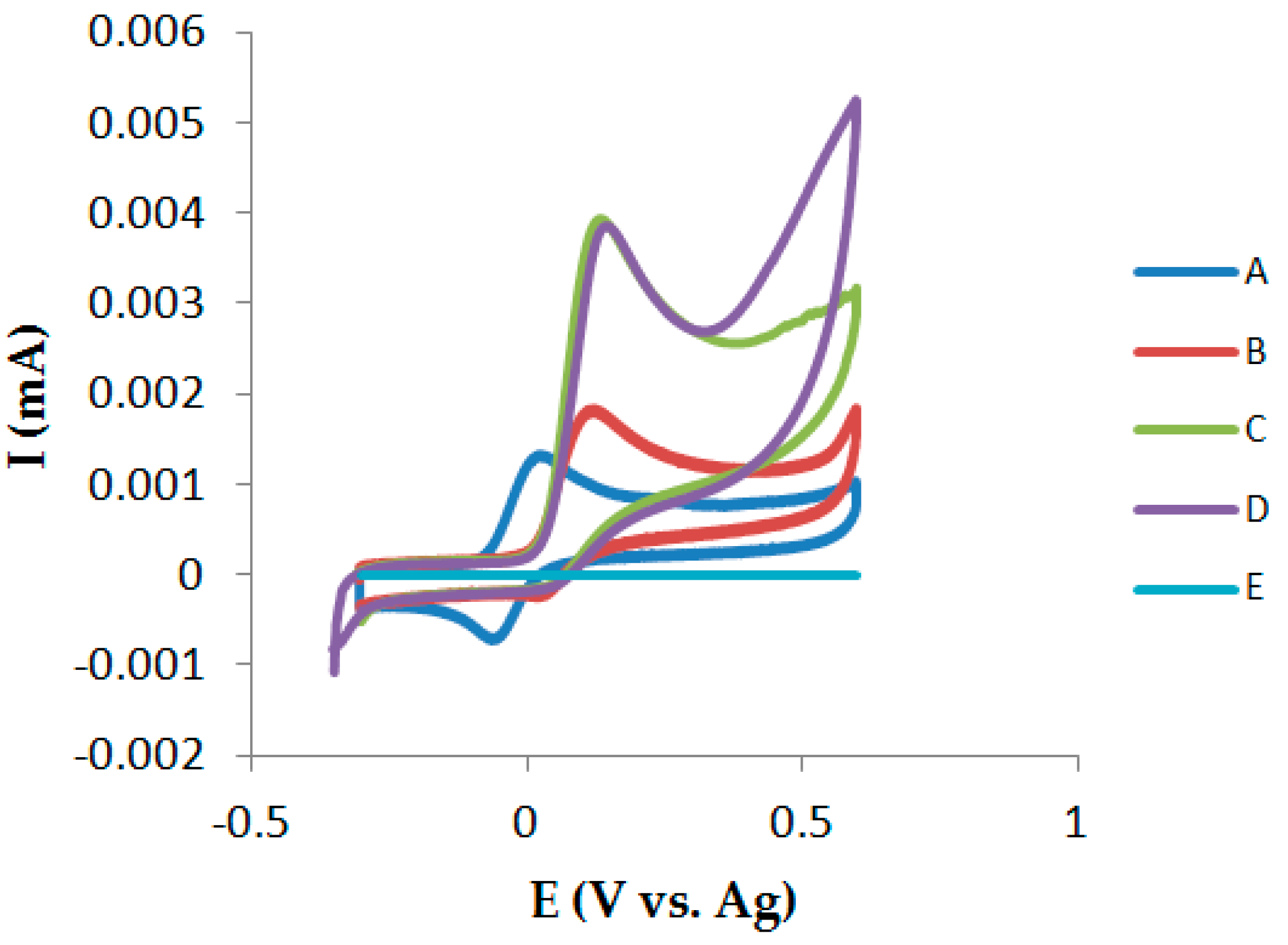
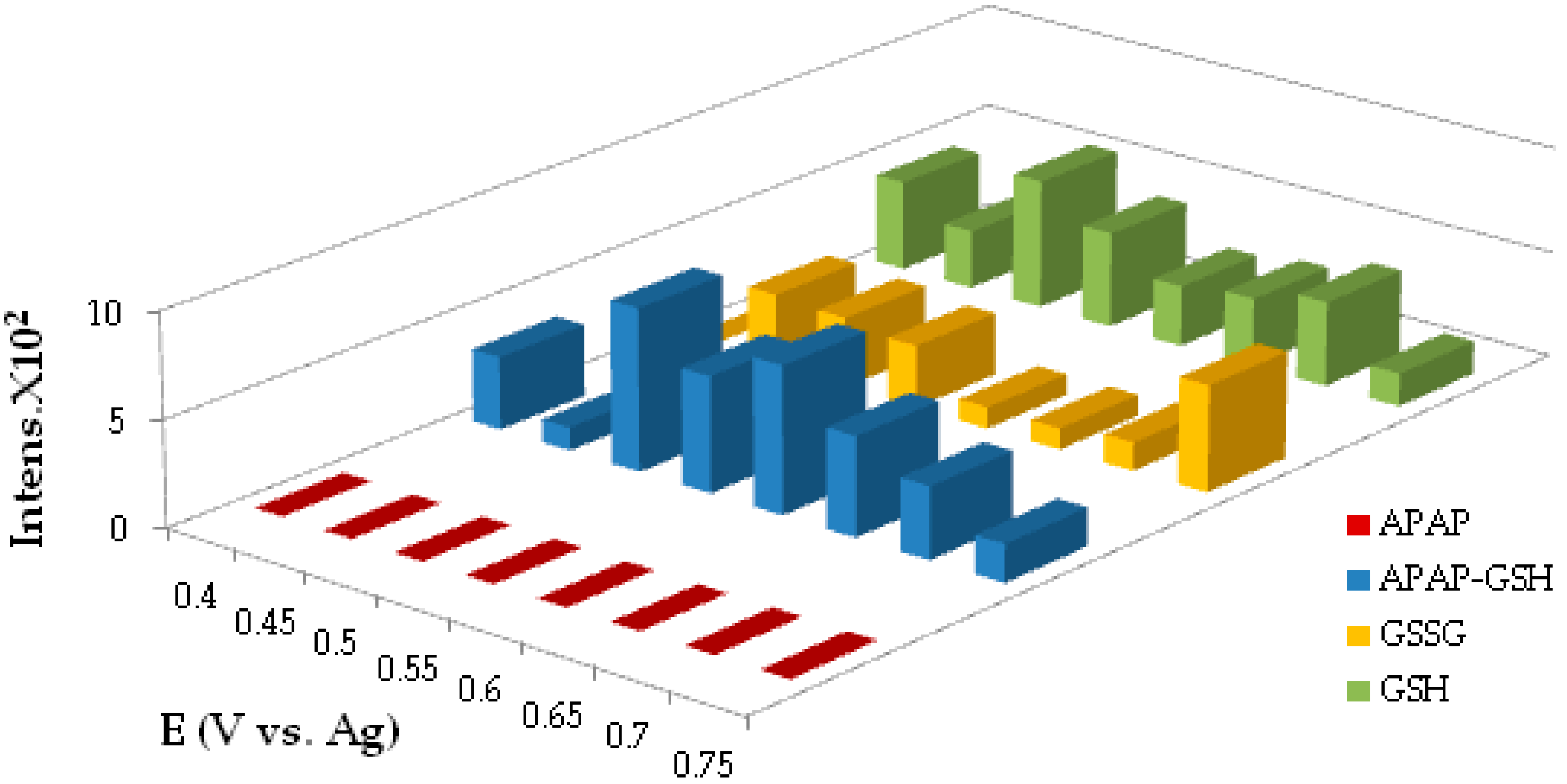
3.4. Cyclic Voltammetry in the Presence of GSH
3.5. Potential Optimization
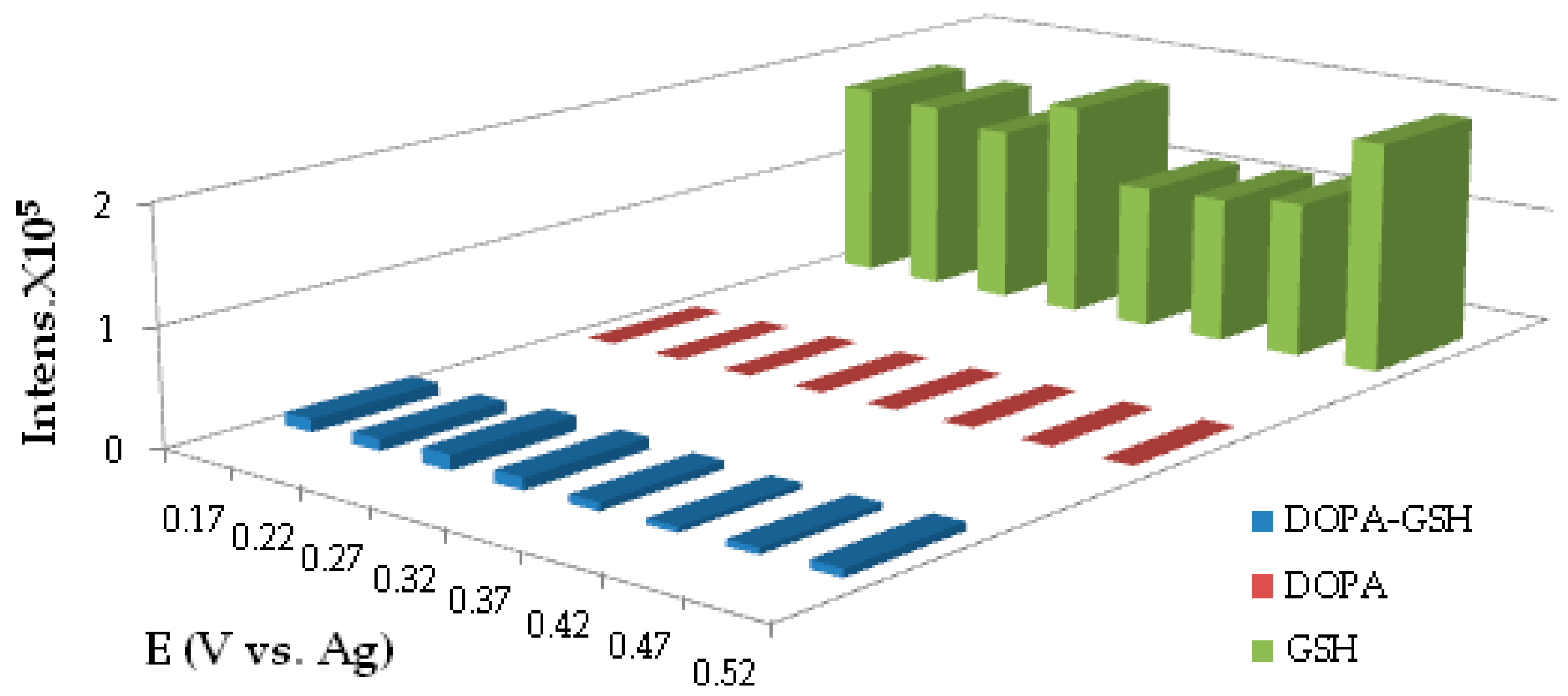
3.6. Exhaustive Electrolysis
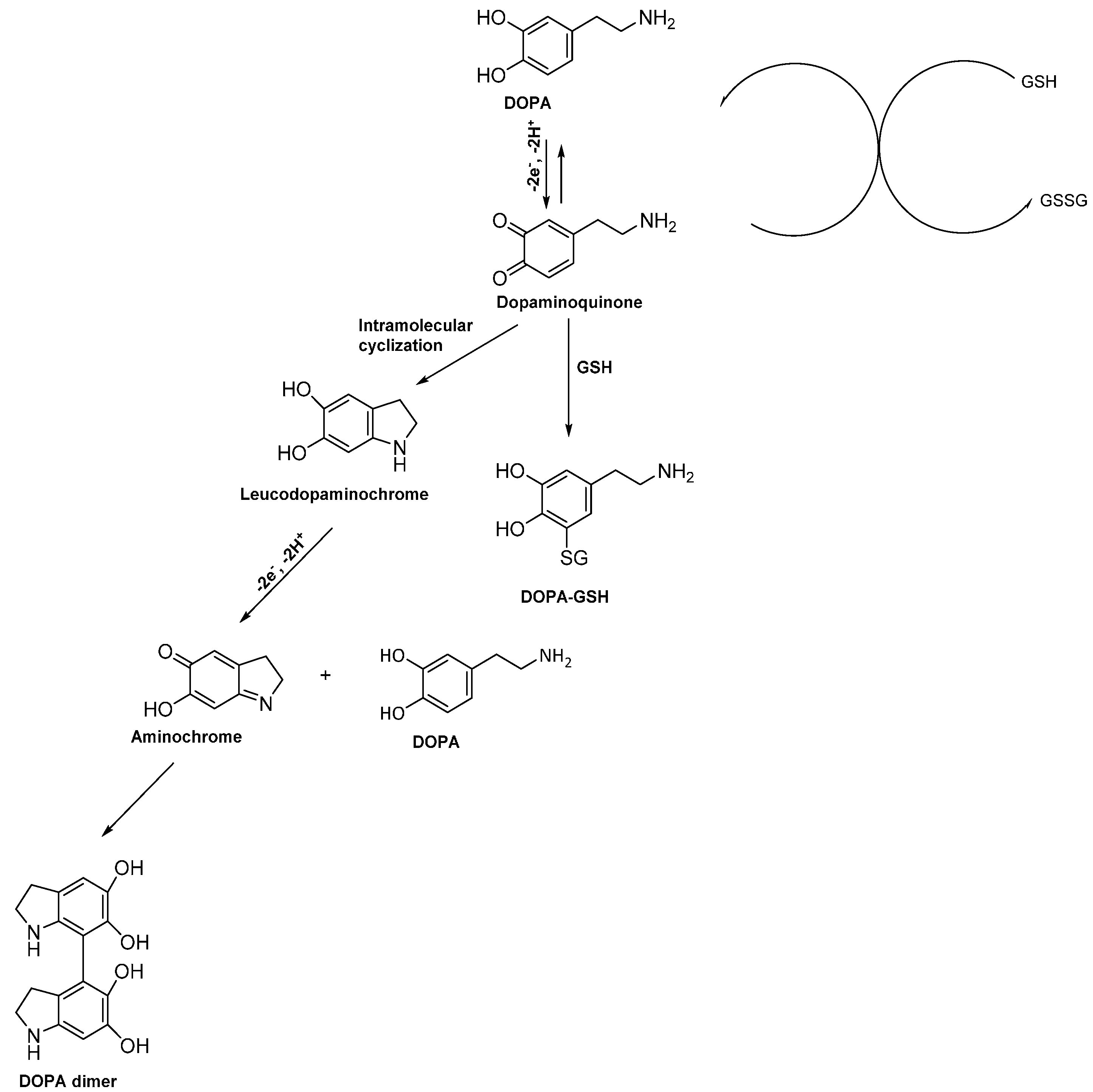
3.7. Proposed Reaction Mechanism for Electrochemical Metabolism
4. Microfluidic Device with an SPE
4.1. Functionality of Chip
4.1.1. Cyclic Voltammetry on Chip
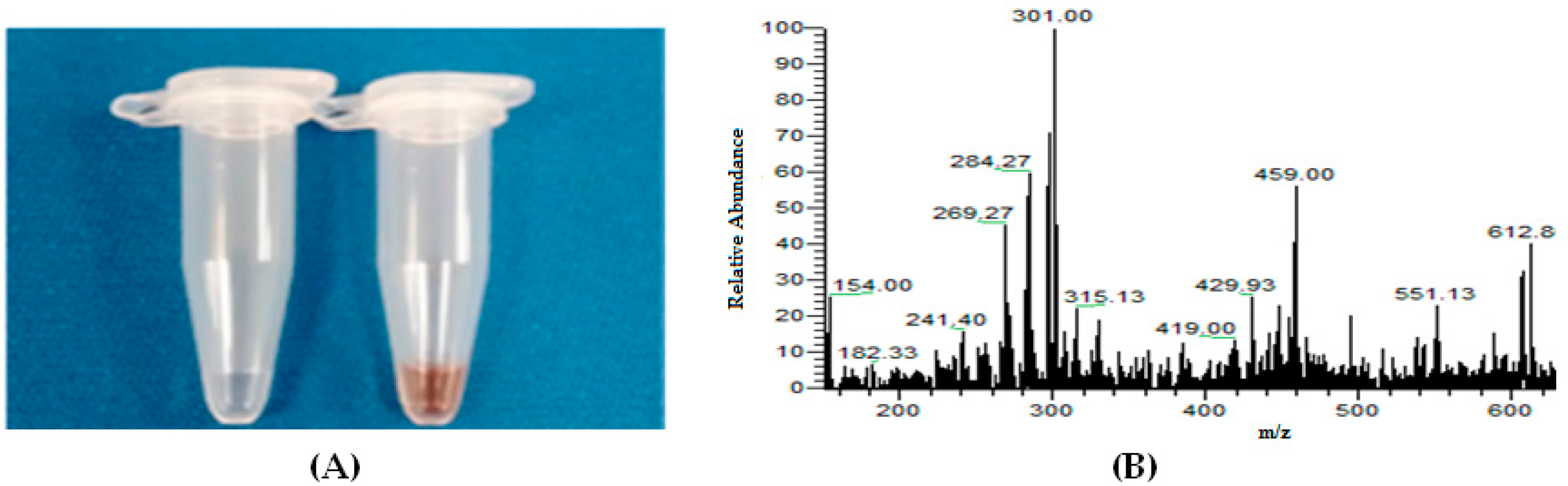
4.1.2. Electrolysis and GSH Conjugation on Chip
4.1.3. Online Coupling
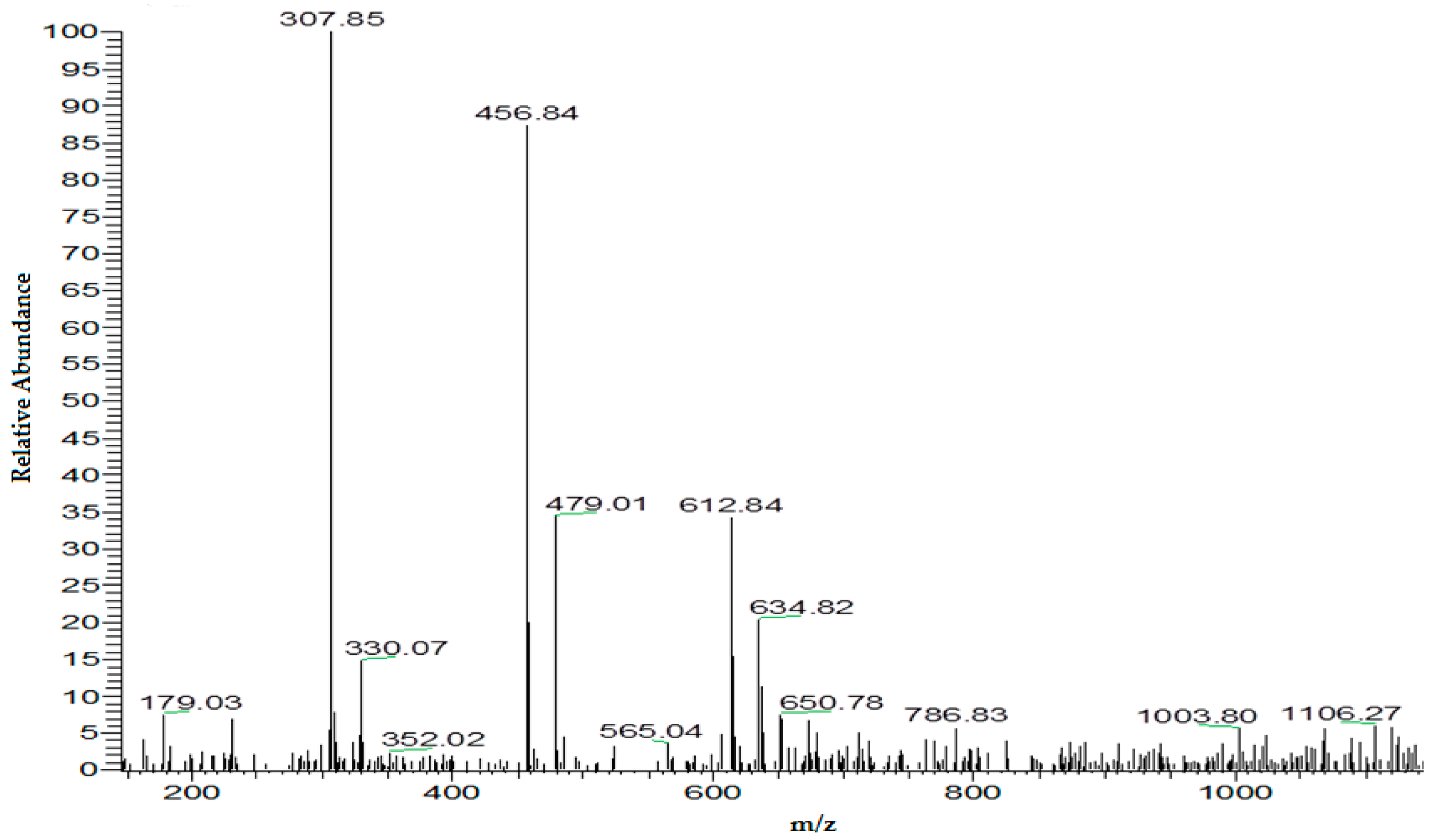
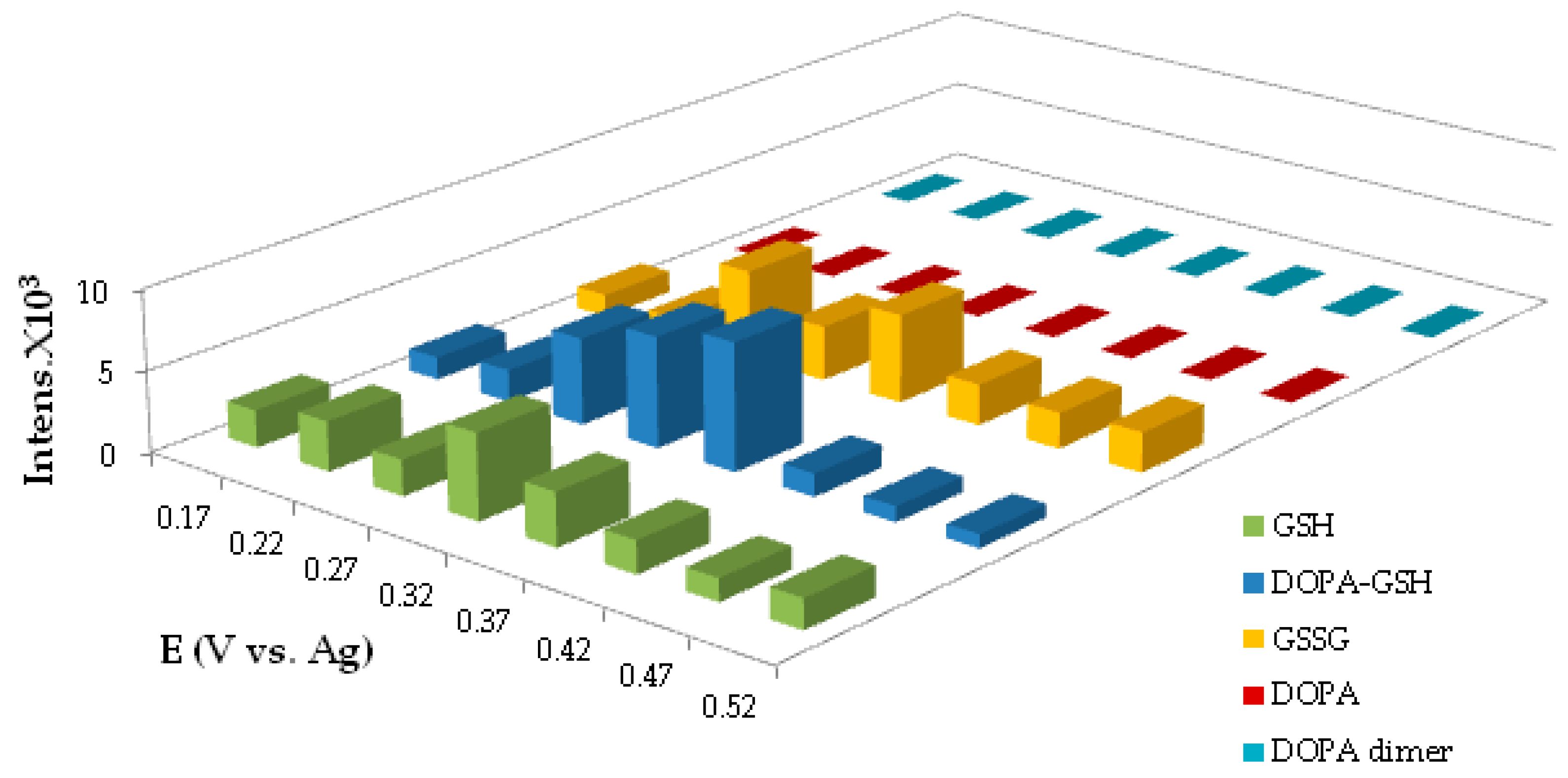
4.1.4. Potential Optimization for the Generation of DOPA-GSH Adduct
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, C.; Li, Y.T.C.; Kong, A.N.T. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: Implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar] [PubMed]
- Jancova, P.; Anzenbacker, P.; Anzenbacherova, E. Phase II metabolizing enzymes. Biomed. Pap. 2010, 154, 103–116. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Alfafara, C.G.; Kanda, A.; Shioi, T.; Shimizu, H.; Shioya, S.; Suga, K. Effect of amino-acids on glutathione production by sacharromyces-cerevisiaie. Appl. Microbiol. Biotechnol. 1992, 36, 538–540. [Google Scholar] [CrossRef]
- Rajasekaran, M.; Abirami, S.; Chen, C. Effects of Single Nucleotide Polymorphisms on Human N-Acetyltransferase 2 Structure and Dynamics by Molecular Dynamics Simulation. PLoS. ONE 2011, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Maggs, J.L.; Antoine, D.J.; Williams, D.P.; Smith, D.A.; Park, B.K. Role of reactive metabolites in drug-induced hepatotoxicity. In Adverse Drug Reactions; Springer: Heidelberg, Germany, 2010; pp. 165–194. [Google Scholar]
- Lohmann, W.; Karst, U. Generation and identification of reactive metabolites by electrochemistry and immobilized enzymes coupled on-line to liquid chromatography/mass spectrometry. Anal. Chem. 2007, 79, 6831–6839. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Karst, U. Online electrochemistry/mass spectrometry in drug metabolism studies: Principles and applications. Expert Opin. Drug Metab. Toxicol. 2010, 6, 715–731. [Google Scholar]
- Van Leeuwen, S.M.; Blankert, B.; Kauffmann, J.M.; Karst, U. Prediction of clozapine metabolism by on-line electrochemistry/liquid chromatography/mass spectrometry. Anal. Bioanal. Chem. 2005, 382, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Plattner, S.; Pitter, F.; Brouwer, H.J.; Oberacher, H. Formation and Characterization of Covalent Guanosine Adducts with Electrochemistry—Liquid Chromatography–Mass Spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 883–884, 198–2014. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, W.; Hayen, H.; Karst, U. Covalent Protein Modification by Reactive Drug Metabolites Using Online Electrochemistry/Liquid Chromatography/Mass Spectrometry. Anal. Chem. 2008, 80, 9714–9719. [Google Scholar] [CrossRef] [PubMed]
- Regino, M.C.S; Brajiter-Toth, A. An electrochemical cell for on-line electrochemistry/mass spectrometry. Anal. Chem. 1997, 69, 5067–5072. [Google Scholar] [CrossRef]
- Getek, T.A.; Korfmacher, W.A.; Mcrae, T.A.; Hinson, J.A. Utility of solution electrochemistry mass spectrometry for investigating the formation and detection of biologically important conjugates of acetaminophen. J. Chromatogr. A 1989, 479, 245–256. [Google Scholar] [CrossRef]
- Lohmann, W.; Karst, U. Simulation of detoxification of paracetamol using on-line electrochemistry/liquid chromatography/mass spectrometry. Anal. Bioanal. Chem. 2006, 386, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Van den Brink, F.T.G.; Buter, L.; Odjik, M.; Olthuis, W.; Karst, U.; van den Berg, A. Mass spectrometric detection of short–lived drug metabolites generated in an electrochemical microfluidic chip. Anal. Chem. 2015, 87, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Roussel, C.; Dayon, L.; Lion, L.; Rohner, T.C.; Josserand, J.; Ropssier, J.S.; Jensen, H.; Girault, H.H. Generation of Mass Tags by the Inherent Electrochemistry of Electrospray for Protein Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Miserere, S.; Ledru, S.; Ruille, N.; Criveau, S.; Boujtita, M.; Bedioui, F. Biocompatible carbon-based screen-printed electrodes for the electrochemical detection of nitric oxide. Electrochem. Commun. 2006, 8, 238–244. [Google Scholar] [CrossRef]
- Mistry, K.K.; Layek, K.; Mahapatra, A.; RoyChaudhurib, C.; Sahab, H. A review on amperometric-type immunosensors based on screen-printed electrodes. Analyst 2014, 139, 2289–2311. [Google Scholar] [CrossRef] [PubMed]
- Khaled, E.; Hassan, H.N.A.; Habib, I.H.I.; Metelka, R. Chitosan modified screen-printed carbon electrode for sensitive analysis of heavy metals. Int. J. Electrochem. Sci. 2010, 5, 158–167. [Google Scholar]
- Kauffmann, J.M.; Antwerpen, P.V.; Sarakbi, A.; Feier, B.; Tarik, S.; Aydogmus, Z. Utility of Screen Printed Electrodes for in Vitro Metabolic Stability Assays: Application to Acetaminophen and its Thioconjugates. Electroanalysis 2011, 23, 2643–2650. [Google Scholar] [CrossRef]
- Monoz, P.; Melendez, C.; Paris, I.; Sequra-Aquilar, J. Molecular and neurochemical mechanism dopamine oxidation to 0-quinone in Parkinson disease pathogenesis. In Toxicity and Autophage in Neurodegenerative Disorders; Springer: Basel, Switzerland, 2015; pp. 205–223. [Google Scholar]
- Eklund, J.C.; Alan, B.M.; Alden, J.A.; Gompton, R.G. Perspectives in modern voltammetry: Basic Concepts and Mechanistic Analysis. Adv. Phys. Org. Chem. 1999, 2, 1–120. [Google Scholar]
- Mahanthesha, K.R.; Kumara-Swamy, B.E.; Vasantakumar-Pai, K.; Chandra, U.; Sherigara, B.S. Cyclic Voltammetric Investigations of Dopamine at Alizarin Modified Carbon Paste Electrode. Int. J. Electrochem. Sci. 2010, 5, 1962–1971. [Google Scholar]
- Nematollahi, D.; Shayani-Jam, H.; Alimoradi, M.; Niroomand, S. Electrochemical oxidation of acetaminophen in aqueous solutions: Kinetic evaluation of hydrolysis, hydroxylation and dimerization processes. Electrochim. Acta 2009, 54, 7407–7515. [Google Scholar] [CrossRef]
- Joseph, B.; Justice, J.R. Introduction to in Vivo Voltammetry. In Voltammetry in the Neurosciences; Adams, R.N., Justice, J.B., Eds.; Humana Press: New York, NY, USA, 1987; pp. 15–17. [Google Scholar]
- Bagheri, A.; Hosseini, H. Electrochemistry of raloxifene on a glassy carbon electrode and its determination in pharmaceutical formulation and human plasma. Bioelectrochemistry 2012, 88, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Hossain, M.E.; Mamum, M.A.; Ehsan, M.Q. Study of redox behavior of Cd(II) and interaction of Cd(II) with proline in the aqueous medium using cyclic voltammetry. J Saud. Chem. Soc. 2012, 16, 145–151. [Google Scholar]
- Ndangli, P.M.; Jijana, A.N.; Olowu, R.N.; Mailu, S.N.; Ngece, F.R.; Williams, A.; Waryo, T.T.; Baker, P.G.L. Impedimetric Response of a Label-Free Genosensor Prepared on a 3-Mercaptoprionic Acid Capped Gallium Selenide Nanocrystal Modified Gold Electrode. Int. J. Electrochem. 2011, 6, 1438–1453. [Google Scholar]
- Ahuja, S.; Jespersen, N. Modern Instrumental Analysis; Elsevier: Kidlington, UK, 2006. [Google Scholar]
- Ke, N.J.; Lu, S.H.; Cheng, S.H. A strategy for the determination of dopamine at a bare glassy carbon electrode: p-Phenylenediamine as a nucleophile. Electrochem. Commun. 2006, 8, 1514–1520. [Google Scholar] [CrossRef]
- Liu, C.Y.; Liu, Z.Y.; Peng, R.; Zhong, Z.C. Quasireversible Process of Dopamine on Copper-Nickel Hydroxide Composite/Nitrogen Doped Graphene/Nafion Modified GCE and Its Electrochemical Application. J. Anal. Methods Chem. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Spina, M.B.; Cohen, G. Dopamine turnover and glutathione oxidation: Implications for Parkinson disease. Proc. Natl. Acad. Sci. USA 1989, 86, 1398–1400. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flow Rate (µL·min−1) | Time of Electrolysis (min) | Combined Flow Rate (µL·min−1) | Time of GSH Conjugation (min) |
|---|---|---|---|
| 2.5 | 12.8 | 5.0 | 4.8 |
| 5 | 6.4 | 10 | 2.4 |
| 10 | 3.2 | 20 | 1.2 |
| 20 | 1.6 | 40 | 0.6 |
| 30 | 1.1 | 60 | 0.4 |
| 60 | 0.6 | 100 | 0.2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasiliadou, R.; Nasr Esfahani, M.M.; Brown, N.J.; Welham, K.J. A Disposable Microfluidic Device with a Screen Printed Electrode for Mimicking Phase II Metabolism. Sensors 2016, 16, 1418. https://doi.org/10.3390/s16091418
Vasiliadou R, Nasr Esfahani MM, Brown NJ, Welham KJ. A Disposable Microfluidic Device with a Screen Printed Electrode for Mimicking Phase II Metabolism. Sensors. 2016; 16(9):1418. https://doi.org/10.3390/s16091418
Chicago/Turabian StyleVasiliadou, Rafaela, Mohammad Mehdi Nasr Esfahani, Nathan J. Brown, and Kevin J. Welham. 2016. "A Disposable Microfluidic Device with a Screen Printed Electrode for Mimicking Phase II Metabolism" Sensors 16, no. 9: 1418. https://doi.org/10.3390/s16091418
APA StyleVasiliadou, R., Nasr Esfahani, M. M., Brown, N. J., & Welham, K. J. (2016). A Disposable Microfluidic Device with a Screen Printed Electrode for Mimicking Phase II Metabolism. Sensors, 16(9), 1418. https://doi.org/10.3390/s16091418