
A Guide to Fluorescent Protein FRET Pairs


Abstract
:1. Introduction
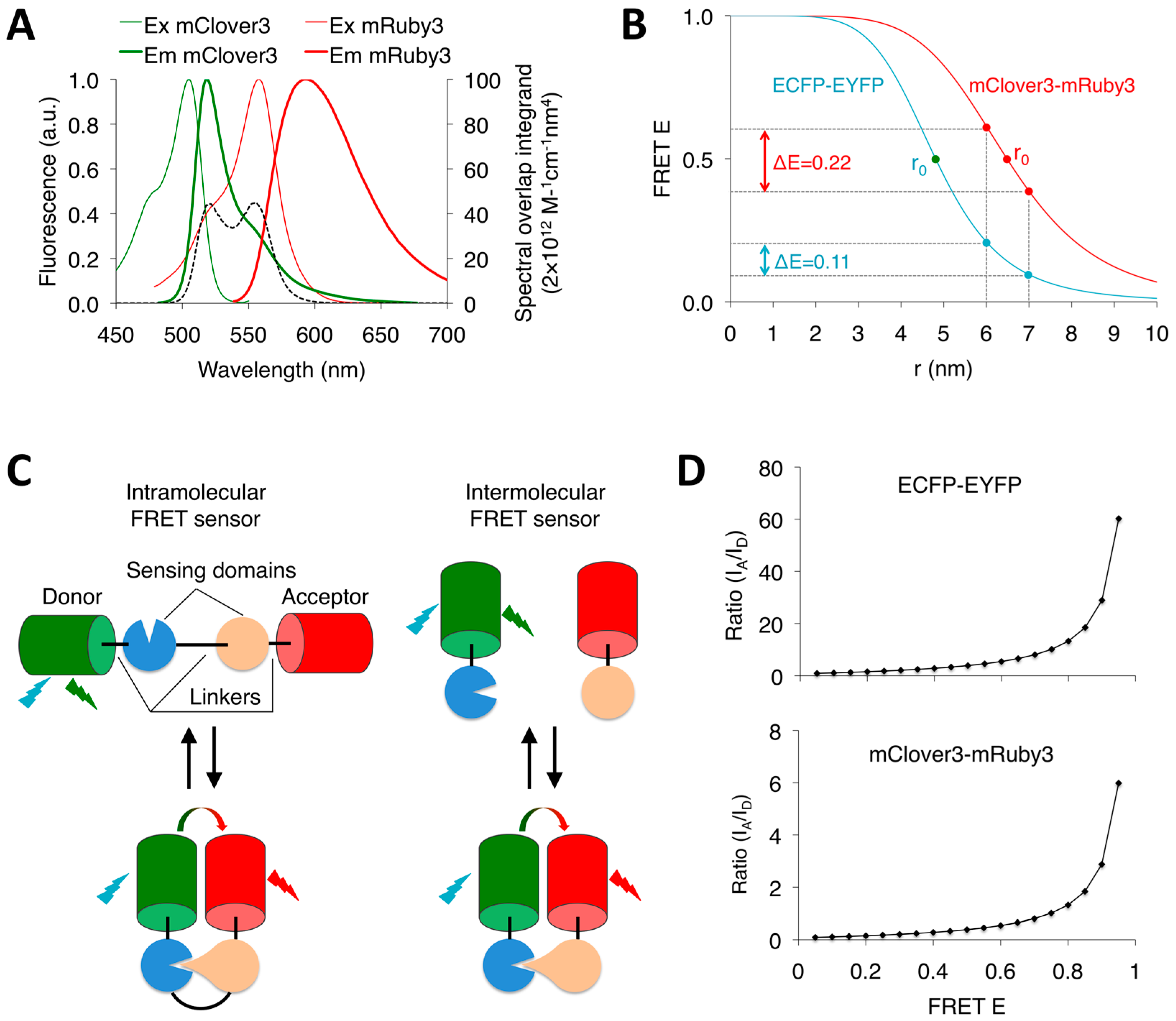
2. FRET Efficiency, FRET Dynamic Range and FRET Measurement
3. Types of FP FRET Pairs
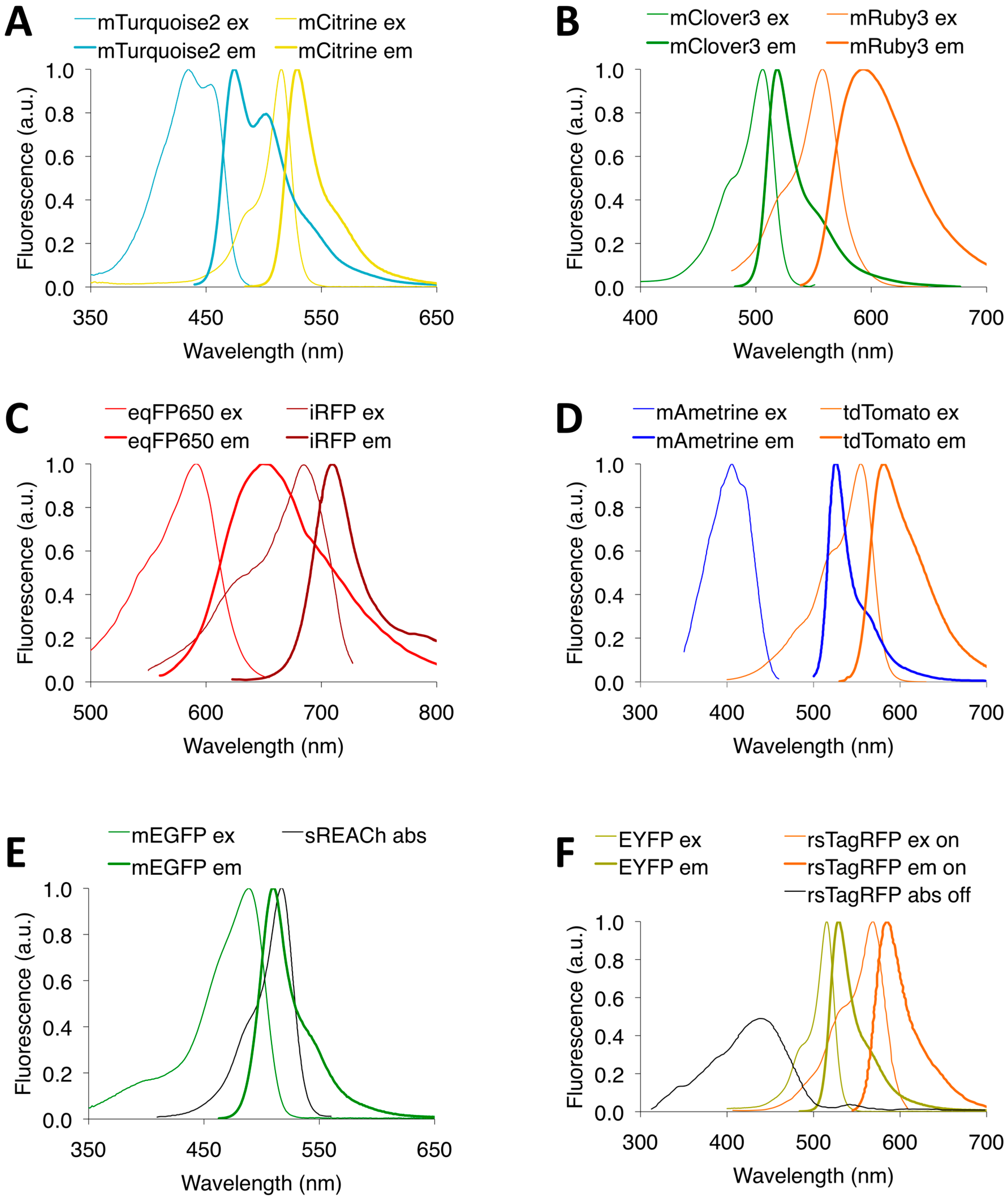
3.1. CFP-YFP FRET Pairs
3.2. GFP-RFP FRET Pairs
3.3. FFP-IFP FRET Pairs
3.4. LSS FP-Based FRET Pairs
3.5. Dark FP-Based FRET Pairs
3.6. Optical Highlighter FP-Based FRET Pairs
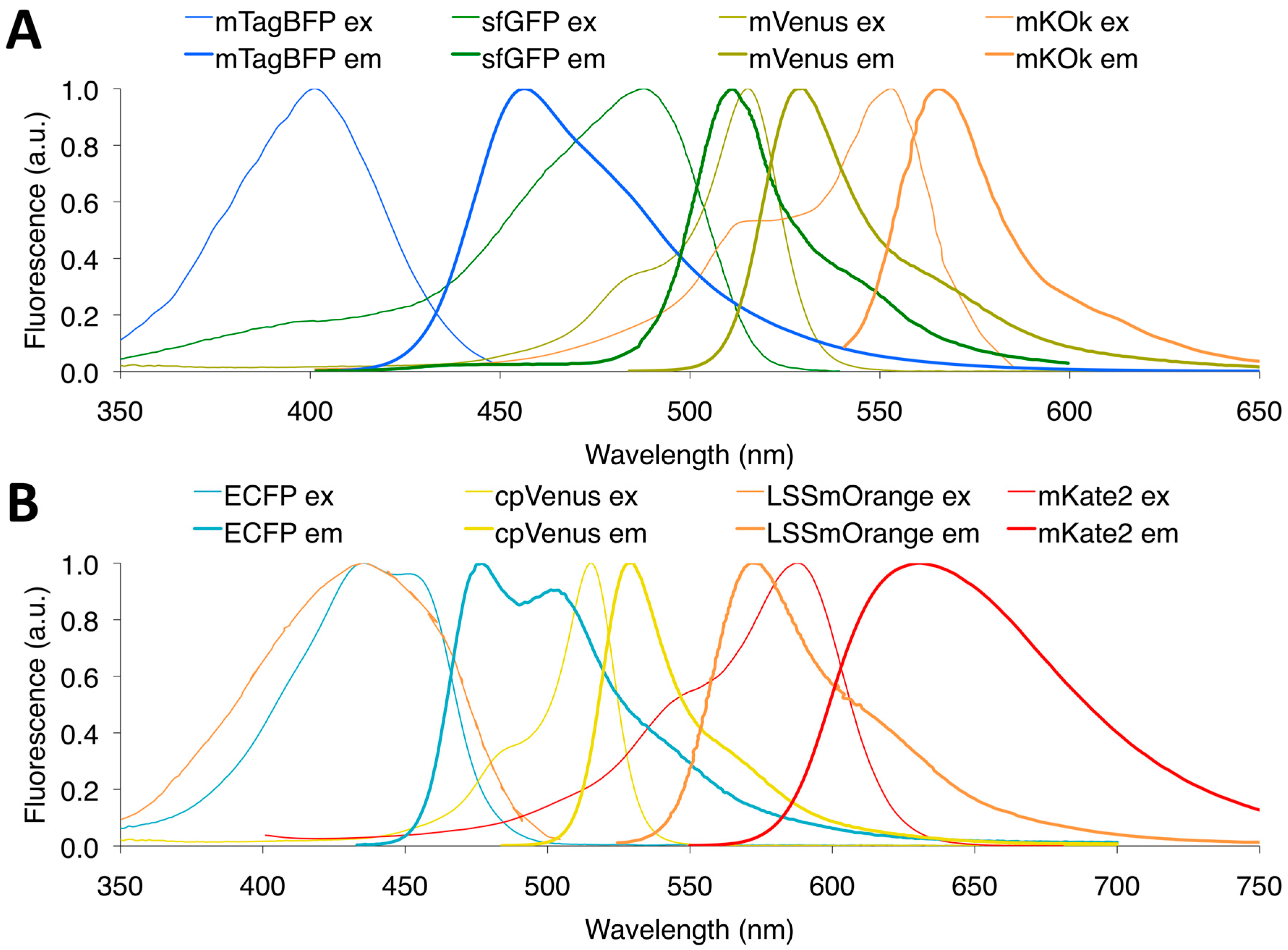
3.7. Multicolor FRET Pairs
3.8. Homo-FRET Pairs
4. Considerations When Using FP Pairs
4.1. FRET Dynamic Range and FRET Change
4.2. Delayed or Decreased on/off Kinetics
4.3. Photostability and pH Sensitivity
4.4. Oligomerization
5. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Forster, T. Energiewanderung und fluoreszenz. Naturwissenschaften 1946, 33, 166–175. [Google Scholar] [CrossRef]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A. Development of probes for cellular functions using fluorescent proteins and fluorescence resonance energy transfer. Annu. Rev. Biochem. 2011, 80, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Masharina, A.; Reymond, L.; Maurel, D.; Umezawa, K.; Johnsson, K. A fluorescent sensor for gaba and synthetic gaba(b) receptor ligands. J. Am. Chem. Soc. 2012, 134, 19026–19034. [Google Scholar] [CrossRef] [PubMed]
- Lesmana, J.; Friedl, P. Destabilization of green fluorescent protein by substitution of its amino-terminal residue. Anim. Cell Technol. Target Mark. 2001, 1, 6–9. [Google Scholar]
- Aoki, K.; Komatsu, N.; Hirata, E.; Kamioka, Y.; Matsuda, M. Stable expression of FRET biosensors: A new light in cancer research. Cancer Sci. 2012, 103, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Use of chimeric fluorescent proteins and fluorescence resonance energy transfer to monitor cellular responses. Circ. Res. 2004, 94, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.S.; Nguyen, T.A.; van der Meer, B.W.; Blank, P.S. The impact of heterogeneity and dark acceptor states on FRET: Implications for using fluorescent protein donors and acceptors. PLoS ONE 2012, 7, e49593. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.L.; Hoppe, A.D. Optimizing fluorescent protein trios for 3-way FRET imaging of protein interactions in living cells. Sci. Rep. 2015, 5, 10270. [Google Scholar] [CrossRef] [PubMed]
- Hoepker, A.C.; Wang, A.; Le Marois, A.; Suhling, K.; Yan, Y.; Marriott, G. Genetically encoded sensors of protein hydrodynamics and molecular proximity. Proc. Natl. Acad. Sci. USA 2015, 112, E2569–E2574. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Tramier, M. FRET microscopy in the living cell: Different approaches, strengths and weaknesses. Bioessays 2012, 34, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Poland, S.P.; Krstajic, N.; Monypenny, J.; Coelho, S.; Tyndall, D.; Walker, R.J.; Devauges, V.; Richardson, J.; Dutton, N.; Barber, P.; et al. A high speed multifocal multiphoton fluorescence lifetime imaging microscope for live-cell FRET imaging. Biomed. Opt. Express 2015, 6, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Rietdorf, J.; Girod, A.; Georget, V.; Pepperkok, R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel gfp2-yfp FRET pair. FEBS Lett. 2002, 531, 245–249. [Google Scholar] [CrossRef]
- Thaler, C.; Koushik, S.V.; Blank, P.S.; Vogel, S.S. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys. J. 2005, 89, 2736–2749. [Google Scholar] [CrossRef] [PubMed]
- Zeug, A.; Woehler, A.; Neher, E.; Ponimaskin, E.G. Quantitative intensity-based FRET approaches—A comparative snapshot. Biophys. J. 2012, 103, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.A.; Rappaz, B.; Webb, D.J.; Brown, C.M. Fluorescence resonance energy transfer microscopy as demonstrated by measuring the activation of the serine/threonine kinase akt. Nat. Protoc. 2013, 8, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Vegh, R.B.; Bravaya, K.B.; Bloch, D.A.; Bommarius, A.S.; Tolbert, L.M.; Verkhovsky, M.; Krylov, A.I.; Solntsev, K.M. Chromophore photoreduction in red fluorescent proteins is responsible for bleaching and phototoxicity. J. Phys. Chem. B 2014, 118, 4527–4534. [Google Scholar] [CrossRef] [PubMed]
- Kirber, M.T.; Chen, K.; Keaney, J.F. Yfp photoconversion revisited: Confirmation of the cfp-like species. Nat. Methods 2007, 4, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Zal, T.; Gascoigne, N.R. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 2004, 86, 3923–3939. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Fluorescence lifetime imaging--techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Pietraszewska-Bogiel, A.; Gadella, T.W. FRET microscopy: From principle to routine technology in cell biology. J. Microsc. 2011, 241, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Day, R.N.; Davidson, M.W. Fluorescent proteins for FRET microscopy: Monitoring protein interactions in living cells. Bioessays 2012, 34, 341–350. [Google Scholar] [CrossRef] [PubMed]
- McGinty, J.; Stuckey, D.W.; Soloviev, V.Y.; Laine, R.; Wylezinska-Arridge, M.; Wells, D.J.; Arridge, S.R.; French, P.M.; Hajnal, J.V.; Sardini, A. In vivo fluorescence lifetime tomography of a FRET probe expressed in mouse. Biomed. Opt. Express 2011, 2, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A.; Christensen, K.; Swanson, J.A. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 2002, 83, 3652–3664. [Google Scholar] [CrossRef]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef]
- Zal, T.; Gascoigne, N.R. Using live FRET imaging to reveal early protein-protein interactions during T cell activation. Curr. Opin. Immunol. 2004, 16, 674–683. [Google Scholar] [PubMed]
- Levitt, J.A.; Matthews, D.R.; Ameer-Beg, S.M.; Suhling, K. Fluorescence lifetime and polarization-resolved imaging in cell biology. Curr. Opin. Biotechnol. 2009, 20, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Carlin, L.M.; Ofo, E.; Barber, P.R.; Vojnovic, B.; Irving, M.; Ng, T.; Ameer-Beg, S.M. Time-lapse FRET microscopy using fluorescence anisotropy. J. Microsc. 2010, 237, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Piston, D.W. High-contrast imaging of fluorescent protein FRET by fluorescence polarization microscopy. Biophys. J. 2005, 88, L14–L16. [Google Scholar] [CrossRef] [PubMed]
- Mattheyses, A.L.; Hoppe, A.D.; Axelrod, D. Polarized fluorescence resonance energy transfer microscopy. Biophys. J. 2004, 87, 2787–2797. [Google Scholar] [CrossRef] [PubMed]
- Kremers, G.J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET forster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Tsien, R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996, 6, 178–182. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef] [PubMed]
- Erard, M.; Fredj, A.; Pasquier, H.; Beltolngar, D.B.; Bousmah, Y.; Derrien, V.; Vincent, P.; Merola, F. Minimum set of mutations needed to optimize cyan fluorescent proteins for live cell imaging. Mol. Biosyst. 2013, 9, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.W.; Henderson, J.N.; Remington, S.J.; Campbell, R.E. Directed evolution of a monomeric, bright and photostable version of clavularia cyan fluorescent protein: Structural characterization and applications in fluorescence imaging. Biochem. J. 2006, 400, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, M.L.; Kremers, G.J.; Kraft, C.A.; Ray, K.; Cranfill, P.J.; Wilson, K.A.; Day, R.N.; Wachter, R.M.; Davidson, M.W.; Rizzo, M.A. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS ONE 2011, 6, e17896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koushik, S.V.; Chen, H.; Thaler, C.; Puhl, H.L.; Vogel, S.S. Cerulean, venus, and venusy67c FRET reference standards. Biophys. J. 2006, 91, L99–L101. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Helmchen, F.; Denk, W. Deep tissue two-photon microscopy. Nat. Methods 2005, 2, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Karasawa, S.; Okamura, Y.; Miyawaki, A. Improving membrane voltage measurements using FRET with new fluorescent proteins. Nat. Methods 2008, 5, 683–685. [Google Scholar] [CrossRef] [PubMed]
- George Abraham, B.; Sarkisyan, K.S.; Mishin, A.S.; Santala, V.; Tkachenko, N.V.; Karp, M. Fluorescent protein based FRET pairs with improved dynamic range for fluorescence lifetime measurements. PLoS ONE 2015, 10, e0134436. [Google Scholar] [CrossRef] [PubMed]
- Sarkisyan, K.S.; Goryashchenko, A.S.; Lidsky, P.V.; Gorbachev, D.A.; Bozhanova, N.G.; Gorokhovatsky, A.Y.; Pereverzeva, A.R.; Ryumina, A.P.; Zherdeva, V.V.; Savitsky, A.P.; et al. Green fluorescent protein with anionic tryptophan-based chromophore and long fluorescence lifetime. Biophys. J. 2015, 109, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016, 6, 20889. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Haynes, R.D.; Corbel, S.Y.; Li, P.; Gonzalez-Gonzalez, E.; Burg, J.S.; Ataie, N.J.; Lam, A.J.; Cranfill, P.J.; Baird, M.A.; et al. Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat. Methods 2014, 11, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filonov, G.S.; Piatkevich, K.D.; Ting, L.M.; Zhang, J.; Kim, K.; Verkhusha, V.V. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 2011, 29, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science 2009, 324, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jackson, W.C.; Steinbach, P.A.; Tsien, R.Y. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc. Natl. Acad. Sci. USA 2004, 101, 16745–16749. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Wang, B.S.; Chen, J.C.; Liu, C.H.; Chou, C.; Chiu, S.J.; Chang, W.Y.; Liu, R.S.; Allen Chang, C.; Lee, Y.J. Mplum-ifp 1.4 fluorescent fusion protein may display forster resonance energy transfer associated properties that can be used for near-infrared based reporter gene imaging. J. Biomed. Opt. 2013, 18, 126013. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, J.; Schnitzer, M.J. An infrared fluorescent protein for deeper imaging. Nat. Biotechnol. 2011, 29, 715–716. [Google Scholar] [CrossRef] [PubMed]
- Zlobovskaya, O.A.; Sarkisyan, K.S.; Lukyanov, K.A. Infrared fluorescent protein irfp as an acceptor for forster resonance energy transfer. Bioorg. Khimiia 2015, 41, 299–304. [Google Scholar] [PubMed]
- Goedhart, J.; van Weeren, L.; Hink, M.A.; Vischer, N.O.; Jalink, K.; Gadella, T.W., Jr. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods 2010, 7, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Rekas, A.; Alattia, J.R.; Nagai, T.; Miyawaki, A.; Ikura, M. Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J. Biol. Chem. 2002, 277, 50573–50578. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Koushik, S.V.; Vogel, S.S.; Gryczynski, I.; Gryczynski, Z. Photophysical properties of cerulean and venus fluorescent proteins. J. Biomed. Opt. 2009, 14, 034047. [Google Scholar] [CrossRef] [PubMed]
- Heikal, A.A.; Hess, S.T.; Baird, G.S.; Tsien, R.Y.; Webb, W.W. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: Coral red (dsred) and yellow (citrine). Proc. Natl. Acad. Sci. USA 2000, 97, 11996–12001. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, B.; Kasper, R.; Seidel, T.; Tinnefeld, P.; Dietz, K.J.; Heilemann, M.; Sauer, M. Fluorescent proteins for single-molecule fluorescence applications. J. Biophotonics 2008, 1, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Shcherbo, D.; Shemiakina, I.I.; Ryabova, A.V.; Luker, K.E.; Schmidt, B.T.; Souslova, E.A.; Gorodnicheva, T.V.; Strukova, L.; Shidlovskiy, K.M.; Britanova, O.V.; et al. Near-infrared fluorescent proteins. Nat. Methods 2010, 7, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.W.; Hazelwood, K.L.; Davidson, M.W.; Campbell, R.E. Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors. Nat. Methods 2008, 5, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, D.M.; Hink, M.A.; Joosen, L.; Gadella, T.W.; Verkhusha, V.V. An orange fluorescent protein with a large stokes shift for single-excitation multicolor fccs and FRET imaging. J. Am. Chem. Soc. 2012, 134, 7913–7923. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lin, M.Z.; McKeown, M.R.; Steinbach, P.A.; Hazelwood, K.L.; Davidson, M.W.; Tsien, R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 2008, 5, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Shibata, A.C.; Nakahata, Y.; Nabekura, J. A dark green fluorescent protein as an acceptor for measurement of forster resonance energy transfer. Sci. Rep. 2015, 5, 15334. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Ameer-Beg, S.M.; Ng, T.T.; Vojnovic, B.; Wouters, F.S. A dark yellow fluorescent protein (yfp)-based resonance energy-accepting chromoprotein (reach) for forster resonance energy transfer with gfp. Proc. Natl. Acad. Sci. USA 2006, 103, 4089–4094. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Zhang, L.; Gadella, T.W.; Gurskaya, N.G.; Lukyanov, K.A.; Verkhusha, V.V. Red fluorescent protein with reversibly photoswitchable absorbance for photochromic FRET. Chem. Biol. 2010, 17, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable gfp for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Don Paul, C.; Kiss, C.; Traore, D.A.; Gong, L.; Wilce, M.C.; Devenish, R.J.; Bradbury, A.; Prescott, M. Phanta: A non-fluorescent photochromic acceptor for pcFRET. PLoS ONE 2013, 8, e75835. [Google Scholar] [CrossRef]
- Zapata-Hommer, O.; Griesbeck, O. Efficiently folding and circularly permuted variants of the sapphire mutant of gfp. BMC Biotechnol. 2003, 3, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subach, O.M.; Gundorov, I.S.; Yoshimura, M.; Subach, F.V.; Zhang, J.; Gruenwald, D.; Souslova, E.A.; Chudakov, D.M.; Verkhusha, V.V. Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 2008, 15, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Oh, Y.; Sens, A.; Ataie, N.; Dana, H.; Macklin, J.J.; Laviv, T.; Welf, E.S.; Dean, K.M.; Zhang, F.; et al. A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat. Biotechnol. 2016, 34, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W.; et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 2007, 4, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Biochemistry, mutagenesis, and oligomerization of dsred, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11984–11989. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Piston, D.W.; Barisas, B.G. Forster distances between green fluorescent protein pairs. Anal. Biochem. 2000, 284, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Shcherbo, D.; Murphy, C.S.; Ermakova, G.V.; Solovieva, E.A.; Chepurnykh, T.V.; Shcheglov, A.S.; Verkhusha, V.V.; Pletnev, V.Z.; Hazelwood, K.L.; Roche, P.M.; et al. Far-red fluorescent tags for protein imaging in living tissues. Biochem. J. 2009, 418, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Lee, S.J.; Yasuda, R. Highly sensitive and quantitative FRET-FLIM imaging in single dendritic spines using improved non-radiative yfp. Brain. Cell Biol. 2008, 36, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Adam, V.; Berardozzi, R.; Byrdin, M.; Bourgeois, D. Phototransformable fluorescent proteins: Future challenges. Curr. Opin. Chem. Biol. 2014, 20, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Demarco, I.A.; Periasamy, A.; Booker, C.F.; Day, R.N. Monitoring dynamic protein interactions with photoquenching FRET. Nat. Methods 2006, 3, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Mizuno, H.; Miyawaki, A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 2004, 306, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Suzuki, T.; Kobayashi, T.; Sakurai, H.; Ohata, H.; Honda, K.; Momose, K.; Namekata, I.; Tanaka, H.; Shigenobu, K.; et al. Simultaneous real-time detection of initiator- and effector-caspase activation by double fluorescence resonance energy transfer analysis. J. Pharmacol. Sci. 2005, 97, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Huang, H.; Shaner, N.C.; Remacle, A.G.; Shiryaev, S.A.; Strongin, A.Y.; Tsien, R.Y.; Wang, Y. Simultaneous visualization of protumorigenic src and mt1-mmp activities with fluorescence resonance energy transfer. Cancer Res. 2010, 70, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.M.; Zhang, W.; McGhee, E.J.; Bunney, T.D.; Talbot, C.B.; Kumar, S.; Munro, I.; Dunsby, C.; Neil, M.A.; Katan, M.; et al. Multiplexed FRET to image multiple signaling events in live cells. Biophys. J. 2008, 95, L69–L71. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Pan, S.; Luo, Q.; Zhang, Z. Monitoring of dual bio-molecular events using FRET biosensors based on mtagbfp/sfgfp and mvenus/mkokappa fluorescent protein pairs. Biosens. Bioelectron. 2013, 46, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Niino, Y.; Hotta, K.; Oka, K. Simultaneous live cell imaging using dual FRET sensors with a single excitation light. PLoS ONE 2009, 4, e6036. [Google Scholar] [CrossRef] [PubMed]
- Galperin, E.; Verkhusha, V.V.; Sorkin, A. Three-chromophore FRET microscopy to analyze multiprotein interactions in living cells. Nat. Methods 2004, 1, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wallrabe, H.; Booker, C.F.; Day, R.N.; Periasamy, A. Three-color spectral FRET microscopy localizes three interacting proteins in living cells. Biophys. J. 2010, 99, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A.D.; Scott, B.L.; Welliver, T.P.; Straight, S.W.; Swanson, J.A. N-way FRET microscopy of multiple protein-protein interactions in live cells. PLoS ONE 2013, 8, e64760. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.; Hanley, Q.S.; Arndt-Jovin, D.J.; Subramaniam, V.; Jovin, T.M. Dynamic fluorescence anisotropy imaging microscopy in the frequency domain (rFLIM). Biophys. J. 2002, 83, 1631–1649. [Google Scholar] [CrossRef]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; en Henegouwen, P.M.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Devauges, V.; Marquer, C.; Lecart, S.; Cossec, J.C.; Potier, M.C.; Fort, E.; Suhling, K.; Leveque-Fort, S. Homodimerization of amyloid precursor protein at the plasma membrane: A homoFRET study by time-resolved fluorescence anisotropy imaging. PLoS ONE 2012, 7, e44434. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.W.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Lindenburg, L.H.; Malisauskas, M.; Sips, T.; van Oppen, L.; Wijnands, S.P.; van de Graaf, S.F.; Merkx, M. Quantifying stickiness: Thermodynamic characterization of intramolecular domain interactions to guide the design of forster resonance energy transfer sensors. Biochemistry 2014, 53, 6370–6381. [Google Scholar] [CrossRef] [PubMed]
- Lindenburg, L.; Merkx, M. Engineering genetically encoded FRET sensors. Sensors 2014, 14, 11691–11713. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, R.; Burnier, J.V.; Ferrar, T.; Beltran-Sastre, V.; Stricher, F.; van der Sloot, A.M.; Garcia-Olivas, R.; Mallabiabarrena, A.; Sanjuan, X.; Zimmermann, T.; et al. Engineering of weak helper interactions for high-efficiency FRET probes. Nat. Methods 2013, 10, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Wriggers, W.; Chakravarty, S.; Jennings, P.A. Control of protein functional dynamics by peptide linkers. Biopolymers 2005, 80, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, K.; Yamada, Y.; Matsuda, T.; Kobayashi, K.; Hashimoto, M.; Matsu-ura, T.; Miyawaki, A.; Michikawa, T.; Mikoshiba, K.; Nagai, T. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow cameleon-nano. Nat. Methods 2010, 7, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Klarenbeek, J.; Goedhart, J.; van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation epac-based FRET sensors for camp feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10, e0122513. [Google Scholar] [CrossRef] [PubMed]
- Lissandron, V.; Terrin, A.; Collini, M.; D’Alfonso, L.; Chirico, G.; Pantano, S.; Zaccolo, M. Improvement of a FRET-based indicator for camp by linker design and stabilization of donor-acceptor interaction. J. Mol. Biol. 2005, 354, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, S.; Spudich, J.A. Systematic control of protein interaction using a modular er/k alpha-helix linker. Proc. Natl. Acad. Sci. USA 2011, 108, 20467–20472. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and gtpases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [PubMed]
- Geiger, A.; Russo, L.; Gensch, T.; Thestrup, T.; Becker, S.; Hopfner, K.P.; Griesinger, C.; Witte, G.; Griesbeck, O. Correlating calcium binding, forster resonance energy transfer, and conformational change in the biosensor tn-xxl. Biophys. J. 2012, 102, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in cdk2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, F.; Marshall, J.D.; Yang, Y.; Gong, Y.; Schnitzer, M.J.; Lin, M.Z. High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat. Neurosci. 2014, 17, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Betolngar, D.B.; Erard, M.; Pasquier, H.; Bousmah, Y.; Diop-Sy, A.; Guiot, E.; Vincent, P.; Merola, F. pH sensitivity of FRET reporters based on cyan and yellow fluorescent proteins. Anal. Bioanal. Chem. 2015, 407, 4183–4193. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Gralle, M.; Dani, M.A.; Lange, D.; Wouters, F.S. Phlameleons: A family of FRET-based protein sensors for quantitative ph imaging. Biochemistry 2008, 47, 13115–13126. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of lipid-modified monomeric gfps into membrane microdomains of live cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from discosoma sp. Red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Kredel, S.; Oswald, F.; Nienhaus, K.; Deuschle, K.; Rocker, C.; Wolff, M.; Heilker, R.; Nienhaus, G.U.; Wiedenmann, J. Mruby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS ONE 2009, 4, e4391. [Google Scholar] [CrossRef] [PubMed]
- Shemiakina, I.I.; Ermakova, G.V.; Cranfill, P.J.; Baird, M.A.; Evans, R.A.; Souslova, E.A.; Staroverov, D.B.; Gorokhovatsky, A.Y.; Putintseva, E.V.; Gorodnicheva, T.V.; et al. A monomeric red fluorescent protein with low cytotoxicity. Nat. Commun. 2012, 3, 1204. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Kwok, S.; Hayashi, Y. Application of FRET probes in the analysis of neuronal plasticity. Front Neural Circuits 2013, 7, 163. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, E.; Aoki, K.; Nakamura, T.; Matsuda, M. Spatiotemporal regulation of small gtpases as revealed by probes based on the principle of forster resonance energy transfer (FRET): Implications for signaling and pharmacology. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, Y. Fluorescence resonance energy transfer biosensors for cancer detection and evaluation of drug efficacy. Clin. Cancer Res. 2010, 16, 3822–3824. [Google Scholar] [CrossRef] [PubMed]
- Bozza, W.P.; Di, X.; Takeda, K.; Rivera Rosado, L.A.; Pariser, S.; Zhang, B. The use of a stably expressed FRET biosensor for determining the potency of cancer drugs. PLoS ONE 2014, 9, e107010. [Google Scholar] [CrossRef] [PubMed]
- Morozova, K.S.; Piatkevich, K.D.; Gould, T.J.; Zhang, J.; Bewersdorf, J.; Verkhusha, V.V. Far-red fluorescent protein excitable with red lasers for flow cytometry and superresolution sted nanoscopy. Biophys. J. 2010, 99, L13–L15. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Escobedo-Lozoya, Y.; Szatmari, E.M.; Yasuda, R. Activation of camkii in single dendritic spines during long-term potentiation. Nature 2009, 458, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.E.; Qin, Y.; Park, J.G.; McCombs, J.E. Design and application of genetically encoded biosensors. Trends Biotechnol. 2011, 29, 144–152. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| siFRET | apFRET | FLIM-FRET | seFRET | prFRET | |
|---|---|---|---|---|---|
| Suitable in live cells | yes | no | yes | yes | yes |
| Temporal resolution | second | no | second * | millisecond | millisecond |
| FRET E change | yes | yes | yes | no | no |
| Fluorescence characteristics | spectrum | intensity | lifetime | intensity | polarization |
| Intramolecular | yes | yes | yes | yes | yes |
| Intermolecular | no | no | yes | yes | yes |
| Control cells | yes | no | yes | yes and no | yes |
| Homo-FRET | no | no | no | no | yes |
| FPs | λex a | λem b | ε c | φ d | BR e | pKa f | Photo-Stability g (min) | Lifetime (ns) | Maturation h (min) | Quaternary Structure | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CFP and YFPs | |||||||||||
| Aquamarine | 430 | 474 | 26 | 0.89 | 23 | 3.3 | 79 | 4.1 | 2 times slower than ECFP i | weak dimer j | [35] |
| ECFP | 433 | 475 | 33 | 0.4 | 13 | 4.7 | 64 | 2.3, 3.0 k | ND | weak dimer j | [40,54] |
| mTurquoise2 | 434 | 474 | 30 | 0.93 | 28 | 3.1 | >64 | 3.8, 4.0 k | ND | monomer | [34] |
| mCerulean3 | 433 | 475 | 40 | 0.8 | 32 | 4.7 | ~35 | 3.7, 3.8 k | Kfold = 1.90 l | monomer | [34,37] |
| LUMP m | 420 | 470 | 24 | 0.55 | 13 | ND | ND | 13.6 | ND | monomer | [11] |
| mTFP1 | 462 | 492 | 64 | 0.85 | 54 | 4.3 | 110 | 3.2 | ND | monomer | [36] |
| EYFP | 513 | 527 | 83 | 0.61 | 51 | 6.9 | 60 | 2.9 | Kfold = 0.39 l | weak dimer j | [40,55] |
| mVenus | 515 | 528 | 92 | 0.57 | 53 | 6 | 15 | 3 | Kfold = 5.62 l | monomer | [55,56] [55,56,57] |
| sEYFP | 515 | 528 | 101 | 0.56 | 57 | 6.9 | ND | ND | ND | weak dimer j | [32] |
| mCitrine | 516 | 529 | 77 | 0.76 | 59 | 5.7 | 49 | 3.61 | ND | monomer | [40,58] |
| YPet | 517 | 530 | 104 | 0.77 | 80 | 5.6 | 49 | ND | ND | dimer | [40] |
| GFPs and RFPs | |||||||||||
| EGFP | 488 | 507 | 56 | 0.6 | 34 | 6 | 174 | 2.4 | 25 | weak dimer j | [40] |
| NowGFP | 494 | 502 | 57 | 0.76 | 43 | 6.2 | ND | 5.1 | ND | monomer | [43] |
| Clover | 505 | 515 | 111 | 0.76 | 84 | 6.1 | 50 | 3 | 30 | weak dimer | [2,46] |
| mClover3 | 506 | 518 | 109 | 0.78 | 85 | 6.5 | 80 | ND | ND | weak dimer | [45] |
| mNeonGreen | 506 | 517 | 116 | 0.8 | 93 | 5.7 | 158 | 3 | 10 | monomer | [46] |
| mRuby2 | 559 | 600 | 113 | 0.38 | 43 | 5.3 | 123 | ND | 150 | monomer | [2] |
| mRuby3 | 558 | 592 | 128 | 0.45 | 58 | 4.8 | 349 | ND | <150 | monomer | [45] |
| mCherry | 587 | 610 | 72 | 0.22 | 16 | 4.5 | 96 | 1.46 | 40 | monomer | [40,59] |
| FFPs and IFPs | |||||||||||
| mPlum | 590 | 649 | 41 | 0.1 | 4 | 4.5 | 53 | ND | 100 | monomer | [40,50] |
| eqFP650 | 592 | 650 | 65 | 0.24 | 16 | 5.7 | 30 n | ND | ND | dimer | [60] |
| mCardinal | 604 | 659 | 87 | 0.19 | 17 | 5.3 | 730 | ND | 27 | weak dimer | [47] |
| IFP1.4 m | 684 | 708 | 92 | 0.07 | 6 | 4.6 | ND | ND | 114 | dimer | [48] |
| iRFP m | 690 | 713 | 105 | 0.06 | 6 | 4 | ND | ND | 168 | dimer | [48] |
| LSS FPs and FP acceptors | |||||||||||
| mAmetrine | 406 | 526 | 45 | 0.58 | 26 | 6 | 2.8 | ND | 48 | monomer | [61,62] |
| LSS-mOrange | 437 | 572 | 52 | 0.45 | 23 | 5.7 | ~2.8 | ND | 138 | monomer | [62] |
| tdTomato | 554 | 581 | 138 | 0.69 | 95 | 4.7 | 98 | 3.1 | 60 | pseudo monomer | [40,63] |
| mKate2 | 588 | 633 | 63 | 0.4 | 25 | 5.4 | 81 | ND | 38 | weak dimer | [47] |
| Dark FPs | |||||||||||
| ShadowG | 486 | 510 | 89 | 0.005 | 0 | ND | ND | ND | 76 | monomer | [64] |
| REACh1 | 495 | 530 | ND | ND | NA | ND | ND | ND | ND | weak dimer | [65] |
| REACh2 | 510 | 538 | ND | ND | NA | ND | ND | ND | ND | weak dimer j | [65] |
| sREACh | 517 | 531 | 115 | 0.07 | 8 | ND | ND | ND | 133 | weak dimer j | [64] |
| Phototransformable FPs | |||||||||||
| rsTagRFP | 440 | 585 | 5 o | 0.005 o | ~0 o | 6.6 | ND | ND | 43 | weak dimer | [66] |
| 15 p | 0.001 p | ~0 p | |||||||||
| 567 | 585 | 37 o | 0.11 o | 4 o | |||||||
| 2 p | 0.11 p | 0.2 p | |||||||||
| PA-GFP | 504 | 517 | 17 | 0.79 | 14 | ND | ND | ND | ND | weak dimer j | [67] |
| Phanta | 506 | 516 | 98 | 0.003 | 0 | 4.5 | ND | ND | ND | monomer | [68] |
| FPs for Multicolor FRET | |||||||||||
| T-Sapphire | 399 | 511 | 44 | 0.6 | 26 | 4.9 | 25 | ND | 78 | weak dimer | [40,69] |
| mTagBFP | 402 | 457 | 52 | 0.63 | 33 | 2.7 | ND | 2.6 | ND | monomer | [70] |
| sfGFP | 485 | 510 | 83 | 0.65 | 54 | 5.5 | 157 | ND | ND | weak dimer j | [46] |
| CyOFP1 | 497 | 589 | 40 | 0.76 | 30.4 | 5.5 | 111 | 3.6 | 15 | weak dimer | [71] |
| mOrange2 | 549 | 565 | 58 | 0.6 | 35 | 6.5 | 228 | ND | 270 | monomer | [63] |
| mKOκ | 551 | 563 | 105 | 0.61 | 64 | 4.2 | ND | ND | ND | monomer | [42] |
| TagRFP | 555 | 584 | 100 | 0.48 | 49 | 3.8 | 37 | 2.3 | 100 | weak dimer | [63,72] |
| DsRed | 556 | 586 | 57 | 0.79 | 45 | 16 | ~678 | 3.65 | ~600 | tetramer | [58,73,74] |
| FRET Pair | φD a | εA (mM−2·cm−1) b | r0 (nm) c | Reference |
|---|---|---|---|---|
| ECFP-EYFP | 0.4 | 83 | 4.9 | [75] |
| mTurquoise2-sEYFP | 0.93 | 101 | 5.9 | [2] |
| mTurquoise2-mVenus | 0.93 | 92 | 5.8 | [35] |
| EGFP-mCherry | 0.6 | 72 | 5.4 | [2] |
| Clover-mRuby2 | 0.76 | 113 | 6.3 | [2] |
| mClover3-mRuby3 | 0.78 | 128 | 6.5 | [45] |
| mNeonGreen-mRuby3 | 0.8 | 128 | 6.5 | [45] |
| eqFP650-iRFP | 0.24 | 105 | 5.8 | this work e |
| mAmetrine-tdTomato d | 0.58 | 138 | 6.6 | this work e |
| LSSmOrange-mKate2 d | 0.45 | 63 | 7.0 | this work e |
| EGFP-sREACh | 0.6 | 115 | 5.8 | [64] |
| EGFP-ShadowG | 0.6 | 89 | 4.7 | [64] |
| EGFP-activated PA-GFP | 0.6 | 17 | 4.4 | this work e |
| EGFP-Phanta | 0.6 | 98 | 5.8 | this work e |
| mTagBFP-sfGFP | 0.63 | 83 | 4.6 | this work e |
| mVenus-mKOκ | 0.57 | 105 | 6.3 | this work e |
| CyOFP1-mCardinal d | 0.76 | 87 | 6.9 | this work e |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16, 1488. https://doi.org/10.3390/s16091488
Bajar BT, Wang ES, Zhang S, Lin MZ, Chu J. A Guide to Fluorescent Protein FRET Pairs. Sensors. 2016; 16(9):1488. https://doi.org/10.3390/s16091488
Chicago/Turabian StyleBajar, Bryce T., Emily S. Wang, Shu Zhang, Michael Z. Lin, and Jun Chu. 2016. "A Guide to Fluorescent Protein FRET Pairs" Sensors 16, no. 9: 1488. https://doi.org/10.3390/s16091488
APA StyleBajar, B. T., Wang, E. S., Zhang, S., Lin, M. Z., & Chu, J. (2016). A Guide to Fluorescent Protein FRET Pairs. Sensors, 16(9), 1488. https://doi.org/10.3390/s16091488