
Development of a PrGO-Modified Electrode for Uric Acid Determination in the Presence of Ascorbic Acid by an Electrochemical Technique


Abstract
:
1. Introduction
2. Experimental
2.1. Reagents and Chemicals
2.2. Preparation of PrGO Modified Electrode
2.3. Instrumentation
3. Results and Discussion
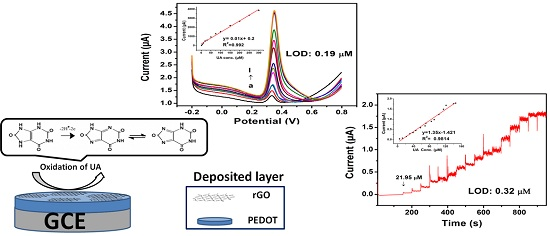
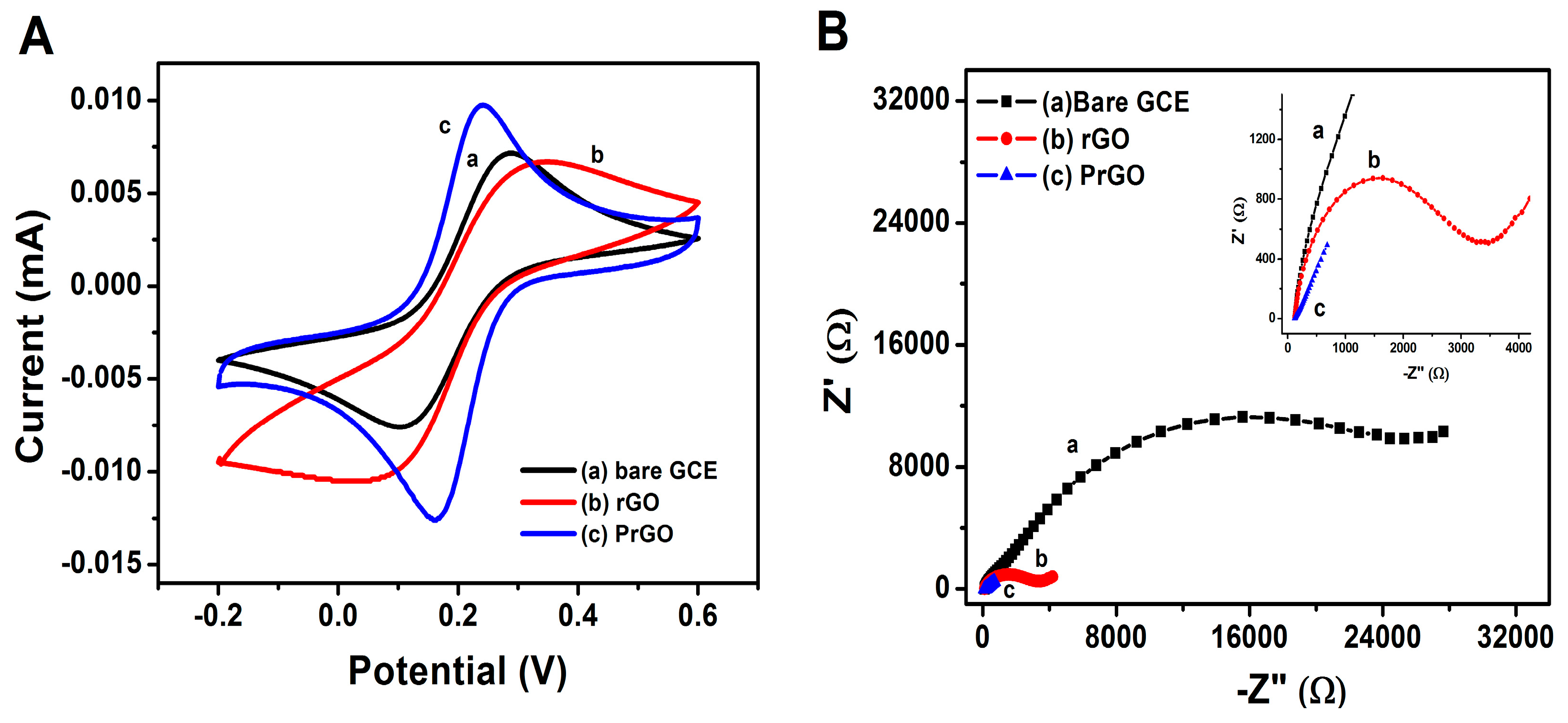
3.1. Electrodeposition
3.2. Material Characterization
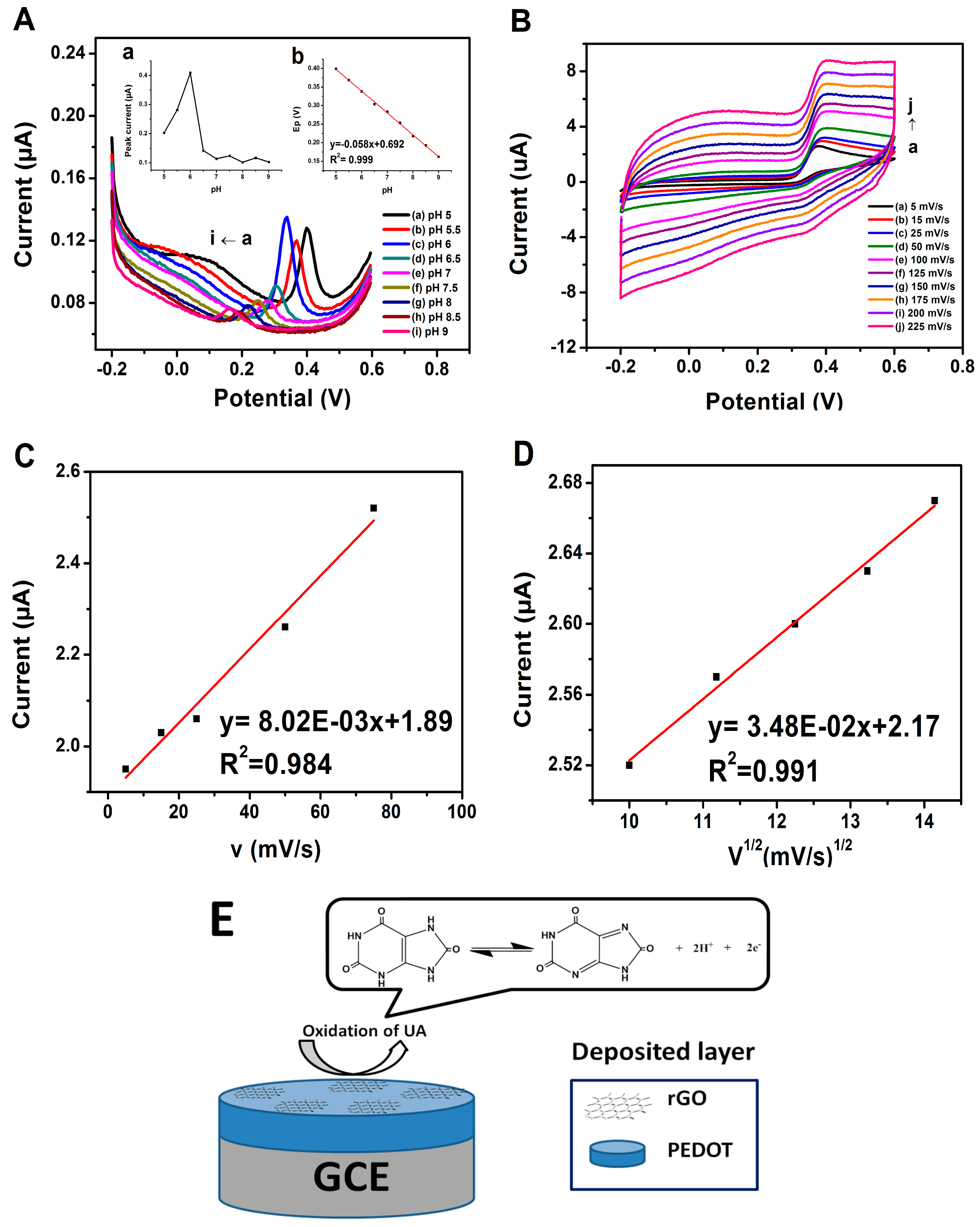
3.3. Effect of pH and Electrode Stability
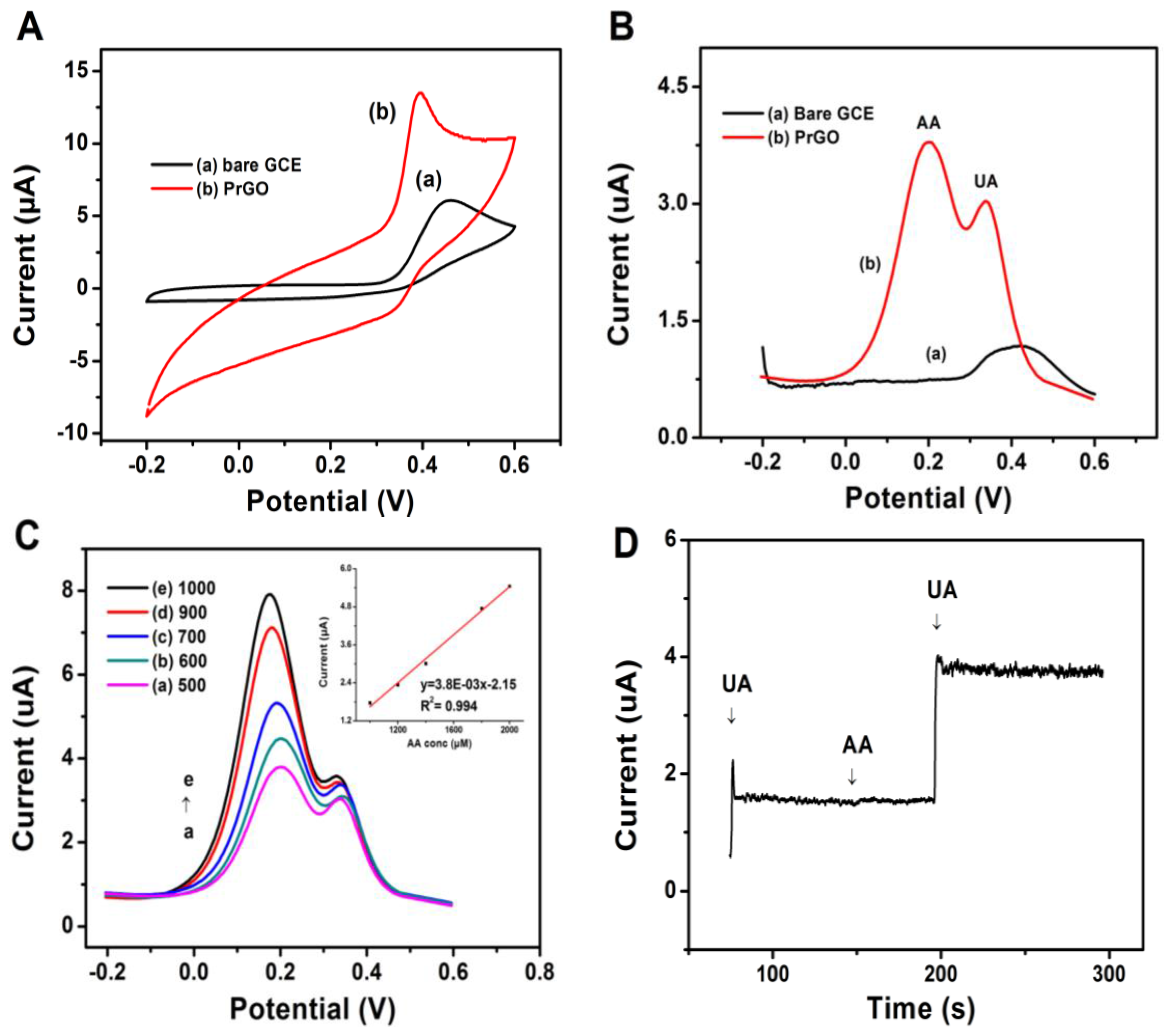
3.4. Oxidation of UA at PrGO
3.5. Limit of Detection
3.6. Analysis of Real Samples
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rock, K.L.; Kataoka, H.; Lai, J.J. Uric acid as a danger signal in gout and its comorbidities. Nat. Rev. Rheumatol. 2013, 9, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Soltani, Z.; Rasheed, K.; Kapusta, D.R.; Reisin, E. Potential role of uric acid in metabolic syndrome, hypertension, kidney injury, and cardiovascular diseases: Is it time for reappraisal? Curr. Hypertens. Rep. 2013, 15, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ma, P.; Wang, A.; Yu, C.; Qian, T.; Wu, S.; Shen, J. Dopamine fluorescent sensors based on polypyrrole/graphene quantum dots core/shell hybrids. Biosens. Bioelectron. 2015, 64, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Hui, N.; Wang, W.; Xu, G.; Luo, X. Graphene oxide doped poly (3,4-ethylenedioxythiophene) modified with copper nanoparticles for high performance nonenzymatic sensing of glucose. J. Mater. Chem. B 2015, 3, 556–561. [Google Scholar] [CrossRef]
- Mousavi, Z.; Bobacka, J.; Lewenstam, A.; Ivaska, A. Response mechanism of potentiometric Ag+ sensor based on poly (3,4-ethylenedioxythiophene) doped with silver hexabromocarborane. J. Electroanal. Chem. 2006, 593, 219–226. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, X.; Zhu, S.; Zhou, Z.; Yao, Y.; Quan, W.; Liu, B. Enhanced sensitivity of ammonia sensor using graphene/polyaniline nanocomposite. Sens. Actuators B Chem. 2013, 178, 485–493. [Google Scholar] [CrossRef]
- Nie, T.; Zhang, O.; Lu, L.; Xu, J.; Wen, Y.; Qiu, X. Facile synthesis of poly (3,4-ethylenedioxythiophene)/graphene nanocomposite and its application for determination of nitrite. Int. J. Electrochem. Sci. 2013, 8, 8708–8718. [Google Scholar]
- Ahammad, A.J.S.; Al Mamun, A.; Akter, T.; Mamun, M.A.; Faraezi, S.; Monira, F.Z. Enzyme-free impedimetric glucose sensor based on gold nanoparticles/polyaniline composite film. J. Solid State Electrochem. 2016, 20, 1933–1939. [Google Scholar] [CrossRef]
- Mondal, S.; Sangaranarayanan, M.V. A novel non-enzymatic sensor for urea using a polypyrrole-coated platinum electrode. Sens. Actuators B Chem. 2013, 177, 478–486. [Google Scholar] [CrossRef]
- Hu, L.; Hecht, D.S.; Gruner, G. Infrared transparent carbon nanotube thin films. Appl. Phys. Lett. 2009, 94, 081103. [Google Scholar] [CrossRef]
- Bhadra, S.; Khastgir, D.; Singha, N.K.; Lee, J.H. Progress in preparation, processing and applications of polyaniline. Prog. Polym. Sci. 2009, 34, 783–810. [Google Scholar] [CrossRef]
- Yamato, H.; Ohwa, M.; Wernet, W. Stability of polypyrrole and poly (3,4-ethylenedioxythiophene) for biosensor application. J. Electroanal. Chem. 1995, 397, 163–170. [Google Scholar] [CrossRef]
- Temmer, R.; Maziz, A.; Plesse, C.; Aabloo, A.; Vidal, F.; Tamm, T. In search of better electroactive polymer actuator materials: Ppy versus pedot versus pedot-ppy composites. Smart Mater. Struct. 2013, 22, 104006. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, S.; Zang, X.; Li, J.; Ma, J. Graphene-based solid-phase extraction combined with flame atomic absorption spectrometry for a sensitive determination of trace amounts of lead in environmental water and vegetable samples. Anal. Chim. Acta 2012, 716, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Pérez-López, B.; Merkoçi, A. Magnetic nanoparticles modified with carbon nanotubes for electrocatalytic magnetoswitchable biosensing applications. Adv. Funct. Mater. 2011, 21, 255–260. [Google Scholar] [CrossRef]
- Samanta, S.K.; Subrahmanyam, K.S.; Bhattacharya, S.; Rao, C.N.R. Composites of graphene and other nanocarbons with organogelators assembled through supramolecular interactions. Chem. Eur. J. 2012, 18, 2890–2901. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hao, Q.; Yang, X.; Lu, L.; Wang, X. A nanostructured graphene/polyaniline hybrid material for supercapacitors. Nanoscale 2010, 2, 2164–2170. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Jung, S.M.; Seo, J.M.; Chang, D.W.; Dai, L.; Baek, J.B. Graphene for energy conversion and storage in fuel cells and supercapacitors. Nano Energy 2012, 1, 534–551. [Google Scholar] [CrossRef]
- Compton, O.C.; Nguyen, S.T. Graphene oxide, highly reduced graphene oxide, and graphene: Versatile building blocks for carbon-based materials. Small 2010, 6, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, Y.; Li, Y.; Feng, H.; Lu, J.; Li, J. Preparation, structure, and electrochemical properties of reduced graphene sheet films. Adv. Funct. Mater. 2009, 19, 2782–2789. [Google Scholar] [CrossRef]
- Li, W.; Liang, C.; Zhou, W.; Qiu, J.; Zhou, Z.; Sun, G.; Xin, Q. Preparation and characterization of multiwalled carbon nanotube-supported platinum for cathode catalysts of direct methanol fuel cells. J. Phys. Chem. B 2003, 107, 6292–6299. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, O.; Xu, J.; Wen, Y.; Duan, X.; Yu, H.; Wu, L.; Nie, T. A facile one-step redox route for the synthesis of graphene/poly (3,4-ethylenedioxythiophene) nanocomposite and their applications in biosensing. Sens. Actuators B Chem. 2013, 181, 567–574. [Google Scholar] [CrossRef]
- Liu, M.; Wen, Y.; Li, D.; He, H.; Xu, J.; Liu, C.; Yue, R.; Lu, B.; Liu, G. Electrochemical immobilization of ascorbate oxidase in poly (3,4-ethylenedioxythiophene)/multiwalled carbon nanotubes composite films. J. Appl. Polym. Sci. 2011, 122, 1142–1151. [Google Scholar] [CrossRef]
- Doğan, H.; Ekinci, D.; Demir, Ã. Atomic scale imaging and spectroscopic characterization of electrochemically reduced graphene oxide. Surf. Sci. 2013, 611, 54–59. [Google Scholar] [CrossRef]
- Guo, H.L.; Wang, X.F.; Qian, Q.Y.; Wang, F.B.; Xia, X.H. A green approach to the synthesis of graphene nanosheets. ACS Nano 2009, 3, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, J. Electrochemical Reactions and Mechanisms in Organic Chemistry; Elsevier: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Schaarschmidt, A.; Farah, A.A.; Aby, A.; Helmy, A.S. Influence of nonadiabatic annealing on the morphology and molecular structure of pedot-pss films. J. Phys. Chem. B 2009, 113, 9352–9355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, X.S. Conducting polymers directly coated on reduced graphene oxide sheets as high-performance supercapacitor electrodes. J. Phys. Chem. C 2012, 116, 5420–5426. [Google Scholar] [CrossRef]
- Choi, K.S.; Liu, F.; Choi, J.S.; Seo, T.S. Fabrication of free-standing multilayered graphene and poly (3,4-ethylenedioxythiophene) composite films with enhanced conductive and mechanical properties. Langmuir 2010, 26, 12902–12908. [Google Scholar] [CrossRef] [PubMed]
- Janata, J.; Josowicz, M. Conducting polymers in electronic chemical sensors. Nat. Mater. 2003, 2, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Pron, A.; Rannou, P. Processible conjugated polymers: From organic semiconductors to organic metals and superconductors. Prog. Polym. Sci. 2002, 27, 135–190. [Google Scholar] [CrossRef]
- Ma, X.; Chao, M.; Wang, Z. Electrochemical detection of dopamine in the presence of epinephrine, uric acid and ascorbic acid using a graphene-modified electrode. Anal. Methods 2012, 4, 1687–1692. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Hu, C.C.; Chu, C.H. Electrochemical impedance characterization of polyaniline-coated graphite electrodes for electrochemical capacitors-effects of film coverage/thickness and anions. J. Electroanal. Chem. 2001, 503, 105–116. [Google Scholar] [CrossRef]
- Shahrokhian, S.; Ghalkhani, M.; Amini, M.K. Application of carbon-paste electrode modified with iron phthalocyanine for voltammetric determination of epinephrine in the presence of ascorbic acid and uric acid. Sens. Actuators B Chem. 2009, 137, 669–675. [Google Scholar] [CrossRef]
- Huang, J.; Liu, Y.; Hou, H.; You, T. Simultaneous electrochemical determination of dopamine, uric acid and ascorbic acid using palladium nanoparticle-loaded carbon nanofibers modified electrode. Biosens. Bioelectron. 2008, 24, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Kamyabi, M.A.; Shafiee, M.A. Electrocatalytic oxidation of dopamine, ascorbic acid and uric acid at poly2, 6 diaminopyridine on the surface of carbon nanotubes/gc electrodes. J. Braz. Chem. Soc. 2012, 23, 593–601. [Google Scholar]
- Yu, L.; Zhang, G.; Wu, Y.; Bai, X.; Guo, D. Cupric oxide nanoflowers synthesized with a simple solution route and their field emission. J. Cryst. Growth 2008, 310, 3125–3130. [Google Scholar] [CrossRef]
- Ping, J.; Wu, J.; Wang, Y.; Ying, Y. Simultaneous determination of ascorbic acid, dopamine and uric acid using high-performance screen-printed graphene electrode. Biosens. Bioelectron. 2012, 34, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J. One-pot synthesis of reduced graphene oxide/zinc sulfide nanocomposite at room temperature for simultaneous determination of ascorbic acid, dopamine and uric acid. Sens. Actuators B Chem. 2015, 221, 750–759. [Google Scholar] [CrossRef]
- Sharath Shankar, S.; Kumara Swamy, B.E.; Chandra, U.; Manjunatha, J.G.; Sherigara, B.S. Simultaneous determination of dopamine, uric acid and ascorbic acid with ctab modified carbon paste electrode. Int. J. Electrochem. Sci. 2009, 4, 592–601. [Google Scholar]
- Dryhurst, G. Electrochemical oxidation of uric acid and xanthine at the pyrolytic graphite electrode mechanistic interpretation of electrochemistry. J. Electrochem. Soc. 1972, 119, 1659–1664. [Google Scholar] [CrossRef]
- Roy, P.R.; Okajima, T.; Ohsaka, T. Simultaneous electroanalysis of dopamine and ascorbic acid using poly (N,N-dimethylaniline)-modified electrodes. Bioelectrochemistry 2003, 59, 11–19. [Google Scholar] [CrossRef]
- Roy, P.R.; Okajima, T.; Ohsaka, T. Simultaneous electrochemical detection of uric acid and ascorbic acid at a poly (N,N-dimethylaniline) film-coated gc electrode. J. Electroanal. Chem. 2004, 561, 75–82. [Google Scholar] [CrossRef]
- Wu, L.; Feng, L.; Ren, J.; Qu, X. Electrochemical detection of dopamine using porphyrin-functionalized graphene. Biosens. Bioelectron. 2012, 34, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fu, H.; Deng, L.; Wang, J. Redox-active thionine-graphene oxide hybrid nanosheet: One-pot, rapid synthesis, and application as a sensing platform for uric acid. Anal. Chim. Acta 2013, 761, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yue, R.; Yao, Z.; Jiang, F.; Du, Y.; Yang, P.; Wang, C. Nonenzymatic uric acid electrochemical sensor based on graphene-modified carbon fiber electrode. Colloids Surf. A Physicochem. Eng. Asp. 2013, 419, 94–99. [Google Scholar] [CrossRef]
- Li, Y.; Ran, G.; Yi, W.J.; Luo, H.Q.; Li, N.B. A glassy carbon electrode modified with graphene and poly(acridine red) for sensing uric acid. Microchim. Acta 2012, 178, 115–121. [Google Scholar] [CrossRef]
- Sun, C.L.; Lee, H.H.; Yang, J.M.; Wu, C.C. The simultaneous electrochemical detection of ascorbic acid, dopamine, and uric acid using graphene/size-selected pt nanocomposites. Biosens. Bioelectron. 2011, 26, 3450–3455. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xie, L.; Li, H. Electrochemical biosensor based on reduced graphene oxide and au nanoparticles entrapped in chitosan/silica sol-gel hybrid membranes for determination of dopamine and uric acid. J. Electroanal. Chem. 2012, 682, 158–163. [Google Scholar] [CrossRef]
- Harish, S.; Mathiyarasu, J.; Phani, K.L.N.; Yegnaraman, V. Pedot/palladium composite material: Synthesis, characterization and application to simultaneous determination of dopamine and uric acid. J. Appl. Electrochem. 2008, 38, 1583–1588. [Google Scholar] [CrossRef]
- Xu, T.Q.; Zhang, Q.L.; Zheng, J.N.; Lv, Z.Y.; Wei, J.; Wang, A.J.; Feng, J.J. Simultaneous determination of dopamine and uric acid in the presence of ascorbic acid using pt nanoparticles supported on reduced graphene oxide. Electrochim. Acta 2014, 115, 109–115. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Techniques | Detection Limit (μM) | Linear Range (μM) | Reference |
|---|---|---|---|---|
| GE/CFE | CV | 0.13 | 0.194–49.68 | [49] |
| Graphene-poly(acridine red)/GCE | DPV | 0.30 | 0.8–150 | [50] |
| Graphene/size-selected Pt | CV, DPV | 0.05 | 0.05–11.9 | [51] |
| RGO–AuNPs–CSHMs | DPV | 0.70 | 1–300 | [52] |
| PEDOT/Palladium | DPV | 7.00 | 7–11 | [53] |
| Pt/RGO | CV, DPV | 0.45 | 10.0–130 | [54] |
| PrGO | DPV | 0.19 | 1–300 | This work |
| Sample | Detected (μM) | Added (μM) | Found (μM) | Recovery (%) |
|---|---|---|---|---|
| Urine 1 | 242.80 | 100 | 342.44 | 99.64% |
| Urine 2 | 244.70 | 100 | 346.29 | 101.59% |
| Urine 3 | 1.36 | 160 | 161.33 | 99.98% |
| Urine 4 | 0.16 | 160 | 160.88 | 100.45% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tukimin, N.; Abdullah, J.; Sulaiman, Y. Development of a PrGO-Modified Electrode for Uric Acid Determination in the Presence of Ascorbic Acid by an Electrochemical Technique. Sensors 2017, 17, 1539. https://doi.org/10.3390/s17071539
Tukimin N, Abdullah J, Sulaiman Y. Development of a PrGO-Modified Electrode for Uric Acid Determination in the Presence of Ascorbic Acid by an Electrochemical Technique. Sensors. 2017; 17(7):1539. https://doi.org/10.3390/s17071539
Chicago/Turabian StyleTukimin, Nurulkhalilah, Jaafar Abdullah, and Yusran Sulaiman. 2017. "Development of a PrGO-Modified Electrode for Uric Acid Determination in the Presence of Ascorbic Acid by an Electrochemical Technique" Sensors 17, no. 7: 1539. https://doi.org/10.3390/s17071539
APA StyleTukimin, N., Abdullah, J., & Sulaiman, Y. (2017). Development of a PrGO-Modified Electrode for Uric Acid Determination in the Presence of Ascorbic Acid by an Electrochemical Technique. Sensors, 17(7), 1539. https://doi.org/10.3390/s17071539