Sensitive Genotyping of Foodborne-Associated Human Noroviruses and Hepatitis A Virus Using an Array-Based Platform

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
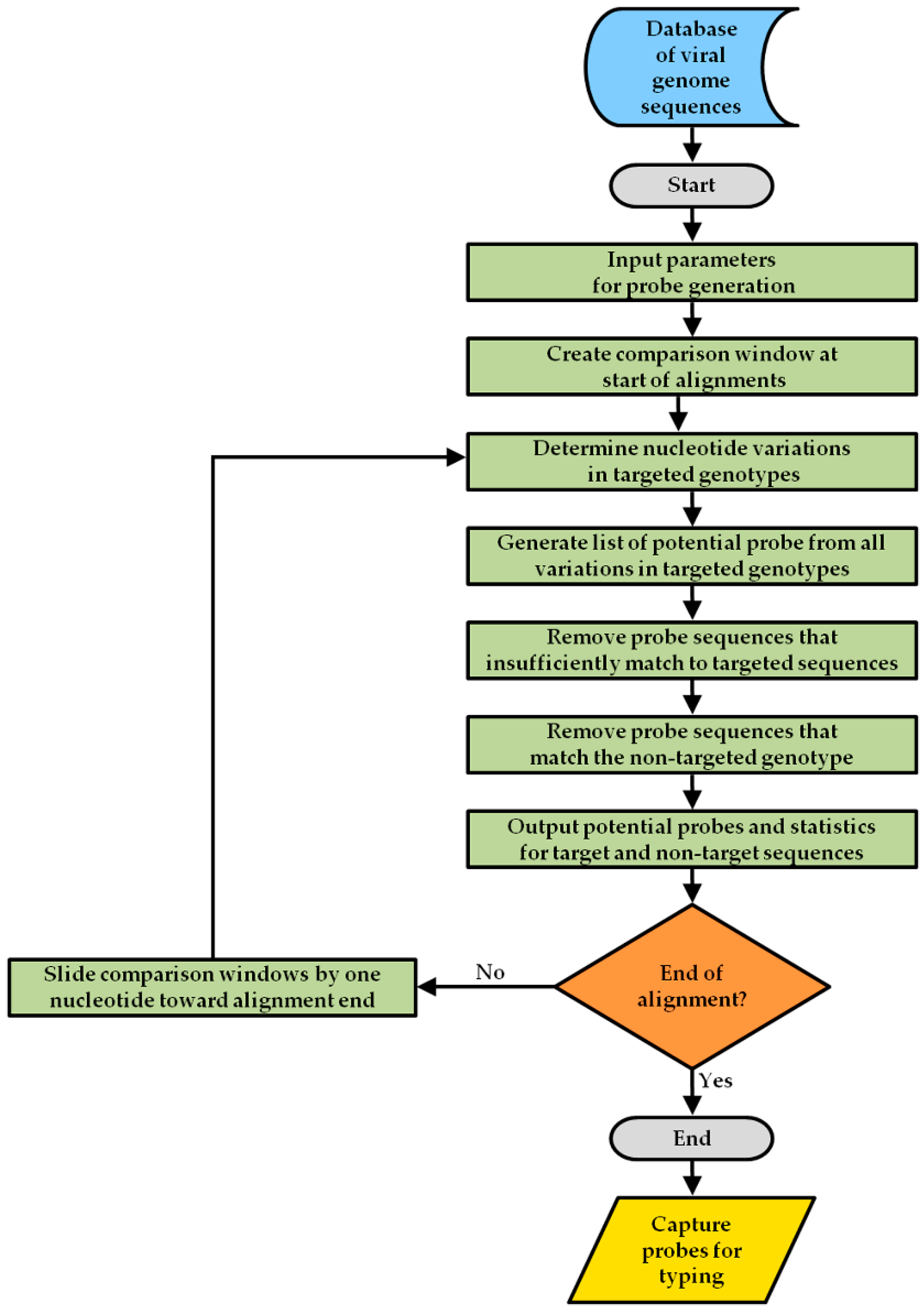
2.1. Design Strategy for the Array Capture Probes
2.2. Spacer Sequence Selection
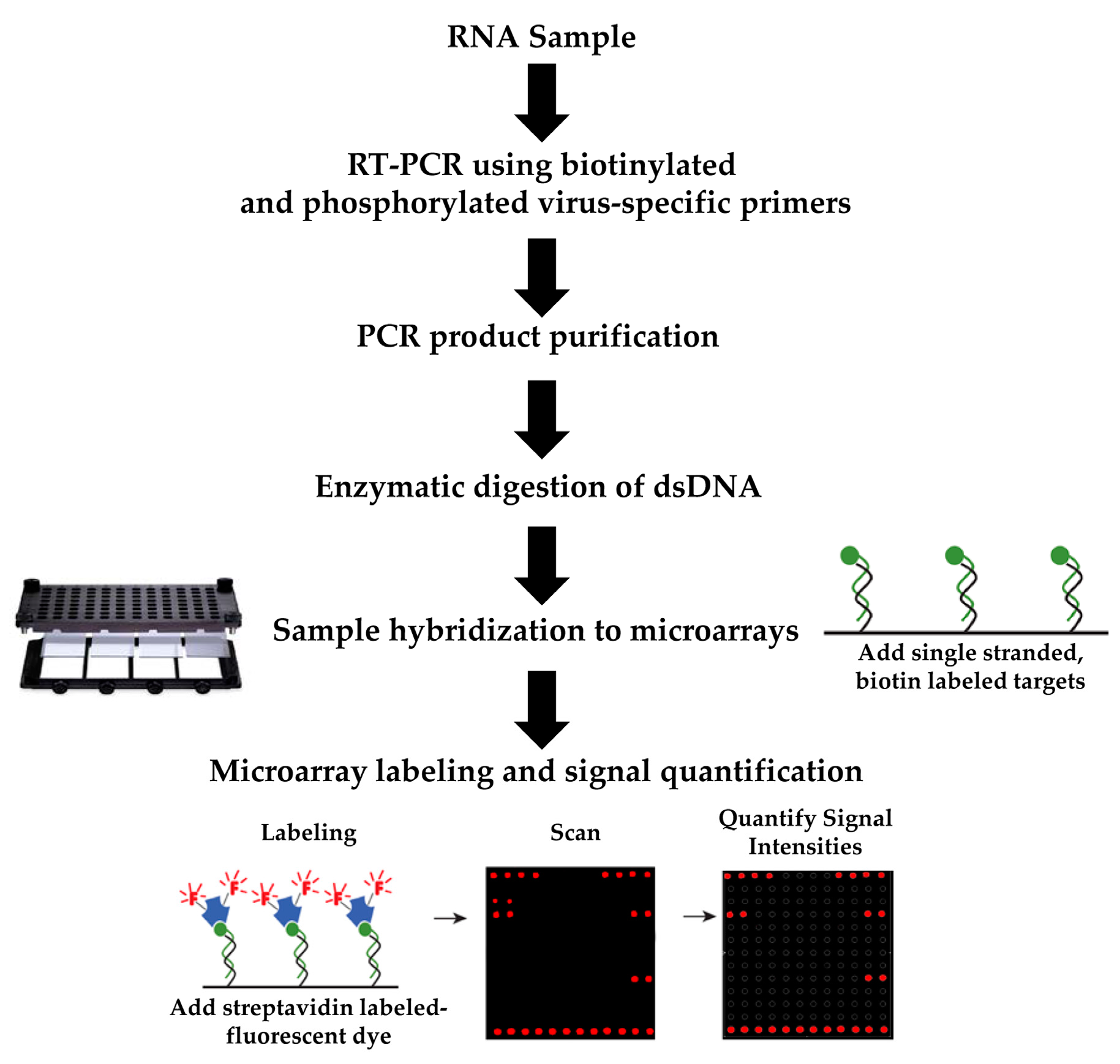
2.3. Virus RNA Sample Preparation and Amplification
2.4. Preparation of In Vitro RNA Transcript Controls
2.5. Microarray Construction and Hybridization
2.6. Microarray Labeling, Signal Amplification, and Data Quantification
3. Results and Discussion
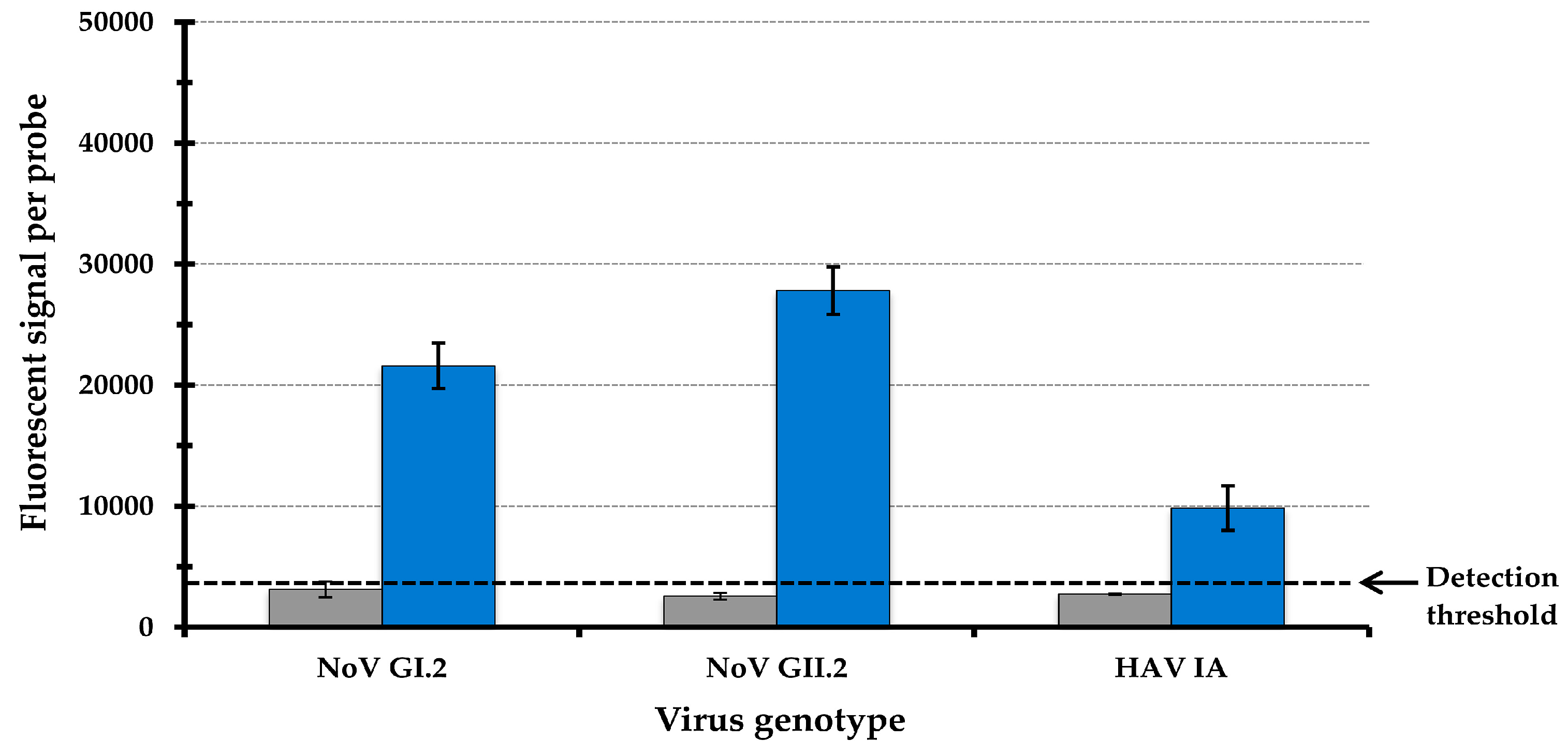
3.1. Validation of Probe Specificity Using the Array-Based Typing Assay for Foodborne Viruses
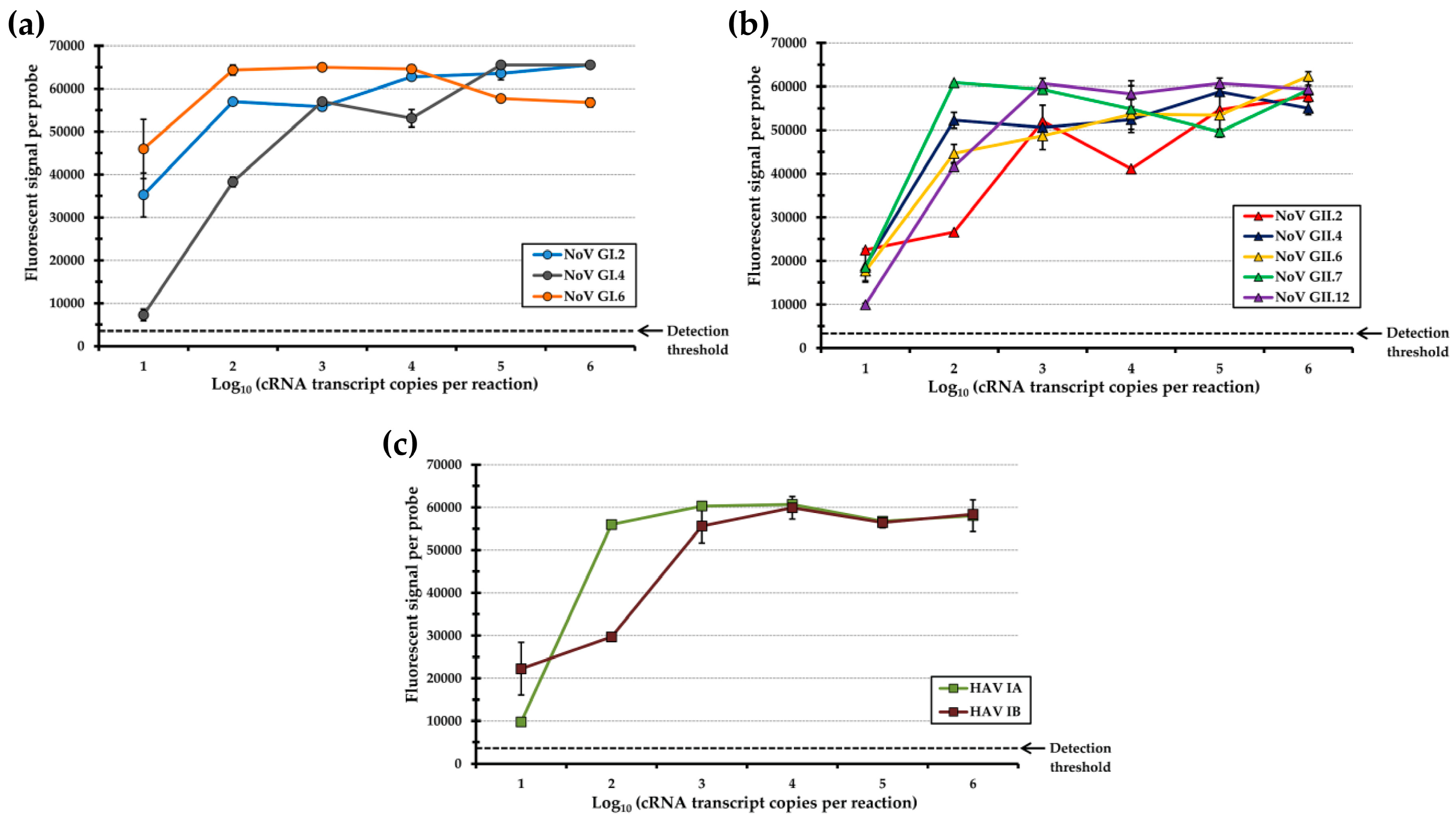
3.2. Inclusion of Spacers in the Capture Probe Sequence Significantly Improved Analytical Sensitivity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall, A.J.; Lopman, B.A.; Payne, D.C.; Patel, M.M.; Gastañaduy, P.A.; Vinjé, J.; Parashar, U.D. Norovirus disease in the United States. Emerg. Infect. Dis. 2013, 19, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Mahon, B.E.; Jones, T.F.; Griffin, P.M. An assessment of the human health impact of seven leading foodborne pathogens in the United States using disability adjusted life years. Epidemiol. Infect. 2015, 143, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Goulter, R.M.; Jaykus, L.A. Human norovirus as a foodborne pathogen: Challenges and developments. Annu. Rev. Food Sci. Technol. 2015, 6, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y. Caliciviridae: The noroviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 582–608. [Google Scholar]
- Robilotti, E.; Deresinski, S.; Pinsky, B.A. Norovirus. Clin. Microbiol. Rev. 2015, 28, 134–164. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J.; Eisenbart, V.G.; Etingue, A.L.; Gould, L.H.; Lopman, B.A.; Parashar, U.D. Epidemiology of foodborne norovirus outbreaks, United States, 2001–2008. Emerg. Infect. Dis. 2012, 18, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Vega, E.; Barclay, L.; Gregoricus, N.; Shirley, S.H.; Lee, D.; Vinjé, J. Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J. Clin. Microbiol. 2014, 52, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Vinjé, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinjé, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. Suppl. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.; Hewitt, J.; Barclay, L.; Ahmed, S.M.; Lake, R.; Hall, A.J.; Lopman, B.; Kroneman, A.; Vennema, H.; Vinjé, J.; et al. Norovirus genotype profiles associated with foodborne transmission, 1999–2012. Emerg. Infect. Dis. 2015, 21, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.; Vennema, H.; van Pelt, W.; Lees, D.; Boshuizen, H.; Henshilwood, K.; Koopmans, M. Use of norovirus genotype profiles to differentiate origins of foodborne outbreaks. Emerg. Infect. Dis. 2010, 16, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Verhoef, L.; Harris, J.; Vennema, H.; Duizer, E.; Van Duynhoven, Y.; Gray, J.; Iturriza, M.; Böttiger, B.; Falkenhorst, G.; et al. Analysis of integrated virological and epidemiological reports of norovirus outbreaks collected within the Foodborne Viruses in Europe network from 1 July 2001 to 30 June 2006. J. Clin. Microbiol. 2008, 46, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, G.; Goncalves Rossi, L.M.; Forbi, J.C.; de Paula, V.S.; Purdy, M.A.; Xia, G.; Khudyakov, Y.E. Hepatitis A virus: Host interactions, molecular epidemiology and evolution. Infect. Genet. Evol. 2014, 21, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Papafragkou, E.; Kulka, M. Review: Approaches to the viral extraction, detection, and identification of hepatitis viruses, HAV and HEV, in foods. J. AOAC Int. 2016, 99, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Collier, M.G.; Tong, X.; Xu, F. Hepatitis A hospitalizations in the United States, 2002–2011. Hepatology 2015, 61, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Calder, L.; Simmons, G.; Thornley, C.; Taylor, P.; Pritchard, K.; Greening, G.; Bishop, J. An outbreak of hepatitis A associated with consumption of raw blueberries. Epidemiol. Infect. 2003, 131, 745–751. [Google Scholar] [CrossRef] [PubMed]
- CDC, Centers for Disease Control and Prevention. 2016—Multistate Outbreak of Hepatitis a Linked to Frozen Strawberries (Final Update). Available online: https://www.cdc.gov/hepatitis/outbreaks/2016/hav-strawberries.htm (accessed on 1 March 2017).
- Collier, M.G.; Khudyakov, Y.E.; Selvage, D.; Adams-Cameron, M.; Epson, E.; Cronquist, A.; Jervis, R.H.; Lamba, K.; Kimura, A.C.; Sowadsky, R.; et al. Outbreak of hepatitis A in the USA associated with frozen pomegranate arils imported from Turkey: An epidemiological case study. Lancet Infect. Dis. 2014, 14, 976–981. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Hepatitis A outbreak associated with green onions at a restaurant—Monaca, Pennsylvania, 2003. MMWR Morb. Mortal. Wkly. Rep. 2003, 52, 1155–1157. [Google Scholar]
- Ramsay, C.N.; Upton, P.A. Hepatitis a and frozen raspberries. Lancet 1989, 1, 43–44. [Google Scholar] [CrossRef]
- Wheeler, C.; Vogt, T.M.; Armstrong, G.L.; Vaughan, G.; Weltman, A.; Nainan, O.V.; Dato, V.; Xia, G.L.; Waller, K.; Amon, J.; et al. An outbreak of hepatitis A associated with green onions. N. Engl. J. Med. 2005, 353, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Nainan, O.V.; Xia, G.; Vaughan, G.; Margolis, H.S. Diagnosis of hepatitis a virus infection: A molecular approach. Clin. Microbiol. Rev. 2006, 19, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Clemens, R.L. The expanding US Market for fresh produce. Iowa Ag Rev. 2015, 10, 8–9. [Google Scholar]
- Huang, S.W. Imports Contribute to Year-Round Fresh Fruit Availability. Fts-356-01. Available online: https://www.ers.usda.gov/webdocs/publications/37056/41739_fts-356-01.pdf?v=41648 (accessed on 9 January 2014).
- Stals, A.; Mathijs, E.; Baert, L.; Botteldoorn, N.; Denayer, S.; Mauroy, A.; Scipioni, A.; Daube, G.; Dierick, K.; Herman, L.; et al. Molecular detection and genotyping of noroviruses. Food Environ. Virol. 2012, 4, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.H.; Jansen, R.W.; Khanna, B.; Totsuka, A.; Nainan, O.V.; Siegl, G.; Widell, A.; Margolis, H.S.; Isomura, S.; Ito, K.; et al. Genetic relatedness of hepatitis A virus strains recovered from different geographical regions. J. Gen. Virol. 1992, 73, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Reddington, K.; Tuite, N.; Minogue, E.; Barry, T. A current overview of commercially available nucleic acid diagnostics approaches to detect and identify human gastroenteritis pathogens. Biomol. Detect. Quantif. 2014, 1, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Heller, R.A.; Theriault, T.P.; Konrad, K.; Lachenmeier, E.; Davis, R.W. Microarrays: Biotechnology’s discovery platform for functional genomics. Trends Biotechnol. 1998, 16, 301–306. [Google Scholar] [CrossRef]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Call, D.R. Challenges and opportunities for pathogen detection using DNA microarrays. Crit. Rev. Microbiol. 2005, 31, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Uttamchandani, M.; Neo, J.L.; Ong, B.N.Z.; Moochhala, S. Applications of microarrays in pathogen detection and biodefence. Trends Biotechnol. 2009, 27, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Law, J.W.-F.; Ab Mutalib, N.-S.; Chan, K.-G.; Lee, L.-H. Rapid methods for the detection of foodborne bacterial pathogens: Principles, applications, advantages and limitations. Front. Microbiol. 2015, 5, 770. [Google Scholar] [CrossRef] [PubMed]
- Ayodeji, M.; Kulka, M.; Jackson, S.A.; Patel, I.; Mammel, M.; Cebula, T.A.; Goswami, B.B. A microarray based approach for the identification of common foodborne viruses. Open Virol. J. 2009, 3, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, N.E.; Fout, G.S. Development and evaluation of a generic tag array to detect and genotype noroviruses in water. J. Virol. Methods 2009, 156, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, X.; Hu, Y.; Yan, H. Reproducibility, fidelity, and discriminant validity of linear RNA amplification for microarray-based identification of major human enteric viruses. Appl. Microbiol. Biotechnol. 2013, 97, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Mammel, M.; Kulka, M.; Patel, I.; Jackson, S.; Goswami, B.B. Detection and identification of common food-borne viruses with a tiling microarray. Open Virol. J. 2011, 5, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, H.; Mammel, M.; Chen, H. Sequence-independent amplification coupled with DNA microarray analysis for detection and genotyping of noroviruses. AMB Express 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jääskeläinen, A.J.; Maunula, L. Applicability of microarray technique for the detection of noro- and astroviruses. J. Virol. Methods 2006, 136, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Mattison, K.; Corneau, N.; Berg, I.; Bosch, A.; Duizer, E.; Gutiérrez-Aguirre, I.; L’Homme, Y.; Lucero, Y.; Luo, Z.; Martyres, A.; et al. Development and validation of a microarray for the confirmation and typing of norovirus RT-PCR products. J. Virol. Methods 2011, 173, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, F.; Corneau, N.; Mattison, K.; Bidawid, S. Development of a DNA microarray for the simultaneous detection and genotyping of noroviruses. J. Food Protect. 2008, 71, 1434–1441. [Google Scholar] [CrossRef]
- Yu, C.; Wales, S.Q.; Mammel, M.K.; Hida, K.; Kulka, M. Optimizing a custom tiling microarray for low input detection and identification of unamplified virus targets. J. Virol. Methods 2016, 234, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Kageyama, T.; Fukushi, S.; Hoshino, F.B.; Shinohara, M.; Uchida, K.; Natori, K.; Takeda, N.; Katayama, K. Genogroup-specific PCR primers for detection of norwalk-like viruses. J. Virol. Methods 2002, 100, 107–114. [Google Scholar] [CrossRef]
- Lu, L.; Ching, K.Z.; de Paula, V.S.; Nakano, T.; Siegl, G.; Weitz, M.; Robertson, B.H. Characterization of the complete genomic sequence of genotype II hepatitis A virus (CF53/Berne isolate). J. Gen. Virol. 2004, 85, 2943–2952. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, B.; Lee, B. Oligonucleotide probes for specific identification of noroviruses and other pathogens. U.S. Patent 20160034636 A1, 2014. [Google Scholar]
- Vega, E.; Barclay, L.; Gregoricus, N.; Williams, K.; Lee, D.; Vinjé, J. Novel surveillance network for norovirus gastroenteritis outbreaks, United States. Emerg. Infect. Dis. 2011, 17, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar]
- Vinjé, J.; Koopmans, M.P. Simultaneous detection and genotyping of “Norwalk-like viruses” by oligonucleotide array in a reverse line blot hybridization format. J. Clin. Microbiol. 2000, 38, 2595–2601. [Google Scholar] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA package 2.0. Algorithm Mol. Biol. 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [PubMed]
- Peplies, J.; Glöckner, F.O.; Amann, R. Optimization strategies for DNA microarray-based detection of bacteria with 16 S rRNA-targeting oligonucleotide probes. Appl. Environ. Microbiol. 2003, 69, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Shchepinov, M.S.; Case-Green, S.C.; Southern, E.M. Steric factors influencing hybridisation of nucleic acids to oligonucleotide arrays. Nucleic Acids Res. 1997, 25, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.; Søe, M.J.; Snakenborg, D.; Møller, L.B.; Dufva, M. Multi-stringency wash of partially hybridized 60-mer probes reveals that the stringency along the probe decreases with distance from the microarray surface. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Gensel, C.L.; Simmons, O.D.; Jaykus, L. Food virology collaborative: NoroCORE tackles foodborne viruses. Food Saf. Mag. 2014, 20, 68–74. [Google Scholar]
- Bialek, S.R.; George, P.A.; Xia, G.L.; Glatzer, M.B.; Motes, M.L.; Veazey, J.E.; Hammond, R.M.; Jones, T.; Shieh, Y.C.; Wamnes, J.; et al. Use of molecular epidemiology to confirm a multistate outbreak of hepatitis A caused by consumption of oysters. Clin. Infect. Dis. 2007, 44, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Forbi, J.C.; Esona, M.D.; Agwale, S.M. Molecular characterization of hepatitis A virus isolates from nigeria. Intervirology 2012, 56, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Hutin, Y.J.F.; Pool, V.; Cramer, E.H.; Nainan, O.V.; Weth, J.; Williams, I.T.; Goldstein, S.T.; Gensheimer, K.F.; Bell, B.P.; Shapiro, C.N.; et al. A multistate, foodborne outbreak of hepatitis A. N. Engl. J. Med. 1999, 340, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Shioda, K.; Barclay, L.; Becker-Dreps, S.; Bucardo-Rivera, F.; Cooper, P.J.; Payne, D.C.; Vinjé, J.; Lopman, B. Can use of viral load improve norovirus clinical diagnosis and disease attribution? Open Forum Infect. Dis. 2017, 4, ofx131. [Google Scholar] [CrossRef]
- Quiñones, B.; Swimley, M.S.; Narm, K.-E.; Patel, R.N.; Cooley, M.B.; Mandrell, R.E. O-antigen and virulence profiling of Shiga toxin-producing Escherichia coli by a rapid and cost-effective DNA microarray colorimetric method. Front. Cell. Infect. Microbiol. 2012, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, B.; Swimley, M.S.; Taylor, A.W.; Dawson, E.D. Identification of Escherichia coli O157 by using a novel colorimetric detection method with DNA microarrays. Foodborne Pathog. Dis. 2011, 8, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.; O’Leary, J.J.; O’Sullivan, N. Real-time reverse transcription PCR detection of norovirus, sapovirus and astrovirus as causative agents of acute viral gastroenteritis. J. Virol. Methods 2007, 146, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, A.A.; McCaustland, K.A.; Zheng, D.-P.; Hadley, L.A.; Vaughn, G.; Adams, S.M.; Ando, T.; Glass, R.I.; Monroe, S.S. Use of TaqMan real-time reverse transcription-PCR for rapid detection, quantification, and typing of norovirus. J. Clin. Microbiol. 2006, 44, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, K.; Huletsky, A.; Peytavi, R.; Turcotte, S.; Veillette, V.; Boissinot, M.; Picard, F.J.; Martel, E.A.; Bergeron, M.G. Rapid exonuclease digestion of PCR-amplified targets for improved microarray hybridization. Clin. Chem. 2007, 53, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, B.; Parker, C.T.; Janda, J.M., Jr.; Miller, W.G.; Mandrell, R.E. Detection and genotyping of Arcobacter and Campylobacter isolates from retail chicken samples by use of DNA oligonucleotide arrays. Appl. Environ. Microbiol. 2007, 73, 3645–3655. [Google Scholar] [CrossRef] [PubMed]
- Haff, R.; Quiñones, B.; Swimley, M.S.; Toyofuku, N. Automatic image analysis and spot classification for detection of pathogenic Escherichia coli on glass slide DNA microarrays. Comput. Electron. Agric. 2010, 71, 163–169. [Google Scholar] [CrossRef]
- Atmar, R.L.; Opekun, A.R.; Gilger, M.A.; Estes, M.K.; Crawford, S.E.; Neill, F.H.; Ramani, S.; Hill, H.; Ferreira, J.; Graham, D.Y. Determination of the 50% human infectious dose for Norwalk virus. J. Infect. Dis. 2014, 209, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Teunis, P.F.M.; Moe, C.L.; Liu, P.; Miller, S.E.; Lindesmith, L.; Baric, R.S.; Le Pendu, J.; Calderon, R.L. Norwalk virus: How infectious is it? J. Med. Virol. 2008, 80, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Halperin, A.; Buhot, A.; Zhulina, E.B. Hybridization at a surface: The role of spacers in DNA microarrays. Langmuir 2006, 22, 11290–11304. [Google Scholar] [CrossRef] [PubMed]
- Coudray-Meunier, C.; Fraisse, A.; Mokhtari, C.; Martin-Latil, S.; Roque-Afonso, A.M. Hepatitis A virus subgenotyping based on RT-qPCR assays. BMC Microbiol. 2014, 14, 296. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.; Kouyos, R.D.; Vennema, H.; Kroneman, A.; Siebenga, J.; van Pelt, W.; Koopmans, M. An integrated approach to identifying international foodborne norovirus outbreaks. Emerg. Infect. Dis. 2011, 17, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Vinjé, J.; Hamidjaja, R.A.; Sobsey, M.D. Development and application of a capsid VP1 (region D) based reverse transcription PCR assay for genotyping of genogroup I and II noroviruses. J. Virol. Methods 2004, 116, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y. Viral pathogen discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Heiden, L. Microarray not fade away. Genet. Eng. News 2014, 34, 1–3. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, J.; Jiang, D.; Xin, Y.; Ding, F.; Deng, Z.; Wang, G.; Ma, X.; Li, F.; Li, G.; et al. A universal oligonucleotide microarray with a minimal number of probes for the detection and identification of viroids at the genus level. PLoS ONE 2013, 8, e64474. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Tested | Genotype | Sample ID | Targeted Genotype by the Capture Probes 1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NoV | HAV | |||||||||||||||
| GI.2 | GI.3 | GI.4 | GI.6 | GI.7 | GII.1 | GII.2 | GII.3 | GII.4 | GII.6 | GII.7 | GII.12 | IA | IB | |||
| NoV | GI.2 | 2012-1 | 59,535 | 475 | 97 | 75 | 83 | 74 | 156 | 346 | 115 | 85 | 94 | 120 | 84 | 106 |
| 2012-2 | 56,624 | 487 | 209 | 131 | 108 | 104 | 176 | 317 | 179 | 346 | 129 | 135 | 112 | 171 | ||
| 2014-1 | 55,381 | 797 | 178 | 164 | 155 | 267 | 188 | 330 | 155 | 164 | 132 | 92 | 91 | 239 | ||
| GI.3 | 2012-3 | 179 | 65,535 | 246 | 279 | 288 | 147 | 211 | 300 | 253 | 163 | 173 | 172 | 171 | 218 | |
| 2012-4 | 198 | 36,025 | 234 | 361 | 229 | 158 | 225 | 319 | 344 | 171 | 198 | 146 | 148 | 253 | ||
| 2014-23 | 248 | 49,256 | 289 | 268 | 238 | 259 | 272 | 438 | 250 | 320 | 321 | 219 | 259 | 274 | ||
| 2014-55 | 234 | 41,789 | 371 | 1291 | 255 | 265 | 304 | 441 | 295 | 424 | 388 | 253 | 289 | 310 | ||
| GI.4 | 2012-5 | 2612 | 469 | 65,535 | 3146 | 171 | 141 | 730 | 399 | 190 | 215 | 198 | 136 | 140 | 200 | |
| 2012-6 | 240 | 371 | 9149 | 449 | 148 | 136 | 311 | 427 | 169 | 230 | 190 | 135 | 134 | 193 | ||
| 2014-35 | 85 | 340 | 58,587 | 670 | 135 | 79 | 212 | 274 | 129 | 105 | 130 | 119 | 103 | 196 | ||
| 2014-58 | 89 | 377 | 46,755 | 486 | 119 | 78 | 179 | 282 | 106 | 145 | 113 | 105 | 95 | 205 | ||
| GI.6 | 2012-7 | 166 | 650 | 674 | 62,158 | 163 | 142 | 252 | 409 | 154 | 351 | 217 | 132 | 133 | 197 | |
| 2012-8 | 673 | 468 | 260 | 65,535 | 152 | 164 | 200 | 295 | 146 | 511 | 209 | 132 | 133 | 165 | ||
| 2014-20 | 134 | 379 | 190 | 65,535 | 360 | 161 | 213 | 324 | 162 | 208 | 183 | 130 | 146 | 193 | ||
| 2014-41 | 137 | 432 | 198 | 65,535 | 355 | 199 | 220 | 346 | 176 | 191 | 188 | 167 | 157 | 216 | ||
| GI.7 | 2014-44 | 199 | 329 | 180 | 176 | 19,726 | 164 | 225 | 288 | 188 | 437 | 260 | 204 | 195 | 224 | |
| GII.1 | 2012-9 | 80 | 274 | 124 | 114 | 106 | 16,444 | 189 | 239 | 108 | 128 | 99 | 153 | 203 | 163 | |
| 2012-10 | 153 | 364 | 192 | 907 | 136 | 65,535 | 247 | 334 | 178 | 183 | 228 | 173 | 130 | 156 | ||
| 2014-18 | 148 | 380 | 172 | 160 | 151 | 24,253 | 248 | 299 | 168 | 163 | 709 | 133 | 242 | 245 | ||
| GII.2 | 2012-11 | 71 | 315 | 79 | 77 | 150 | 172 | 65,535 | 235 | 119 | 99 | 156 | 1562 | 82 | 91 | |
| 2012-12 | 78 | 232 | 75 | 78 | 156 | 87 | 65,535 | 377 | 105 | 308 | 383 | 521 | 87 | 93 | ||
| 2014-48 | 119 | 371 | 154 | 145 | 138 | 125 | 30,397 | 151 | 173 | 132 | 228 | 157 | 138 | 180 | ||
| GII.3 | 2014-31 | 85 | 281 | 80 | 90 | 104 | 86 | 216 | 65,535 | 196 | 113 | 114 | 156 | 90 | 99 | |
| 2014-39 | 78 | 142 | 81 | 90 | 93 | 85 | 211 | 65,535 | 189 | 111 | 115 | 129 | 93 | 102 | ||
| GII.4 New Orleans | 2012-13 | 150 | 331 | 191 | 250 | 137 | 162 | 303 | 203 | 65,535 | 256 | 241 | 161 | 126 | 163 | |
| 2012-14 | 298 | 242 | 336 | 237 | 227 | 211 | 277 | 221 | 55,210 | 245 | 529 | 295 | 284 | 246 | ||
| 2014-2 | 169 | 370 | 181 | 176 | 154 | 139 | 326 | 204 | 33,307 | 159 | 189 | 560 | 157 | 185 | ||
| 2014-3 | 133 | 399 | 198 | 159 | 152 | 141 | 336 | 204 | 46,211 | 182 | 412 | 157 | 219 | 239 | ||
| GII.4 Sydney | 2014-4 | 153 | 401 | 178 | 163 | 146 | 139 | 336 | 230 | 47,929 | 223 | 619 | 294 | 223 | 226 | |
| 2014-14 | 298 | 517 | 206 | 195 | 220 | 181 | 371 | 338 | 65,535 | 305 | 304 | 267 | 305 | 367 | ||
| GII.6 | 2014-12 | 73 | 180 | 75 | 79 | 94 | 72 | 187 | 135 | 218 | 9375 | 85 | 103 | 81 | 79 | |
| 2014-13 | 63 | 236 | 79 | 80 | 133 | 73 | 208 | 332 | 442 | 65,535 | 115 | 103 | 81 | 93 | ||
| GII.7 | 2014-30 | 197 | 388 | 63 | 110 | 112 | 69 | 468 | 264 | 104 | 87 | 58,415 | 170 | 90 | 205 | |
| 2014-54 | 287 | 376 | 130 | 128 | 122 | 79 | 2158 | 296 | 598 | 116 | 60,589 | 677 | 101 | 252 | ||
| GII.12 | 2012-16 | 167 | 367 | 182 | 647 | 150 | 232 | 350 | 309 | 167 | 664 | 208 | 65,535 | 124 | 147 | |
| 2014-6 | 145 | 399 | 176 | 145 | 134 | 123 | 324 | 196 | 197 | 364 | 1496 | 15,745 | 128 | 174 | ||
| 2014-43 | 138 | 358 | 170 | 157 | 134 | 124 | 290 | 186 | 145 | 139 | 673 | 11,201 | 134 | 160 | ||
| HAV | IA | 2014-3 | 211 | 444 | 242 | 216 | 206 | 217 | 311 | 333 | 232 | 207 | 297 | 213 | 14,067 | 270 |
| 2014-5 | 166 | 326 | 227 | 207 | 185 | 181 | 325 | 252 | 210 | 185 | 181 | 189 | 8073 | 222 | ||
| 2014-6 | 190 | 417 | 240 | 236 | 191 | 190 | 325 | 305 | 204 | 267 | 228 | 169 | 6370 | 235 | ||
| IB | 1347 | 204 | 432 | 235 | 221 | 174 | 178 | 315 | 268 | 208 | 190 | 191 | 170 | 312 | 4479 | |
| 1357 | 229 | 453 | 264 | 218 | 207 | 180 | 325 | 309 | 224 | 187 | 194 | 174 | 250 | 41,234 | ||
| 1373 | 203 | 476 | 245 | 230 | 183 | 176 | 370 | 323 | 219 | 182 | 194 | 175 | 214 | 8418 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiñones, B.; Lee, B.G.; Martinsky, T.J.; Yambao, J.C.; Haje, P.K.; Schena, M. Sensitive Genotyping of Foodborne-Associated Human Noroviruses and Hepatitis A Virus Using an Array-Based Platform. Sensors 2017, 17, 2157. https://doi.org/10.3390/s17092157
Quiñones B, Lee BG, Martinsky TJ, Yambao JC, Haje PK, Schena M. Sensitive Genotyping of Foodborne-Associated Human Noroviruses and Hepatitis A Virus Using an Array-Based Platform. Sensors. 2017; 17(9):2157. https://doi.org/10.3390/s17092157
Chicago/Turabian StyleQuiñones, Beatriz, Bertram G. Lee, Todd J. Martinsky, Jaszemyn C. Yambao, Paul K. Haje, and Mark Schena. 2017. "Sensitive Genotyping of Foodborne-Associated Human Noroviruses and Hepatitis A Virus Using an Array-Based Platform" Sensors 17, no. 9: 2157. https://doi.org/10.3390/s17092157
APA StyleQuiñones, B., Lee, B. G., Martinsky, T. J., Yambao, J. C., Haje, P. K., & Schena, M. (2017). Sensitive Genotyping of Foodborne-Associated Human Noroviruses and Hepatitis A Virus Using an Array-Based Platform. Sensors, 17(9), 2157. https://doi.org/10.3390/s17092157