Redox Cycling Realized in Paper-Based Biochemical Sensor for Selective Detection of Reversible Redox Molecules Without Micro/Nano Fabrication Process

Abstract
:1. Introduction
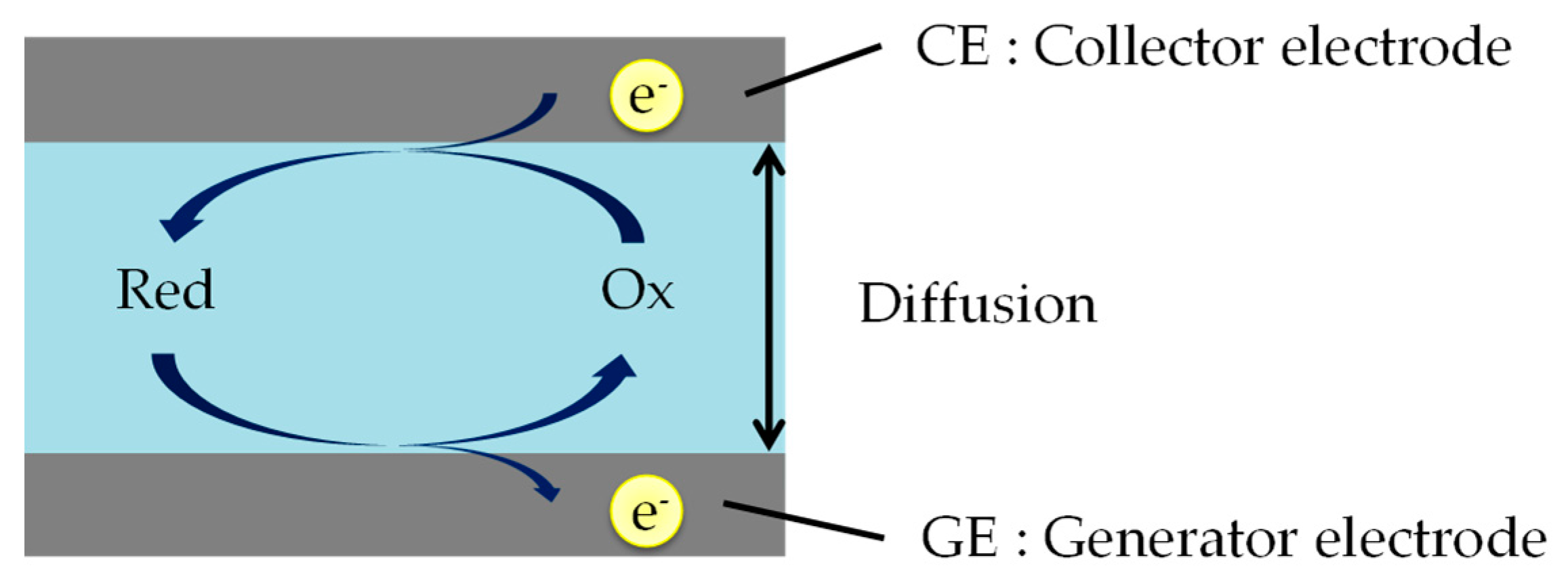
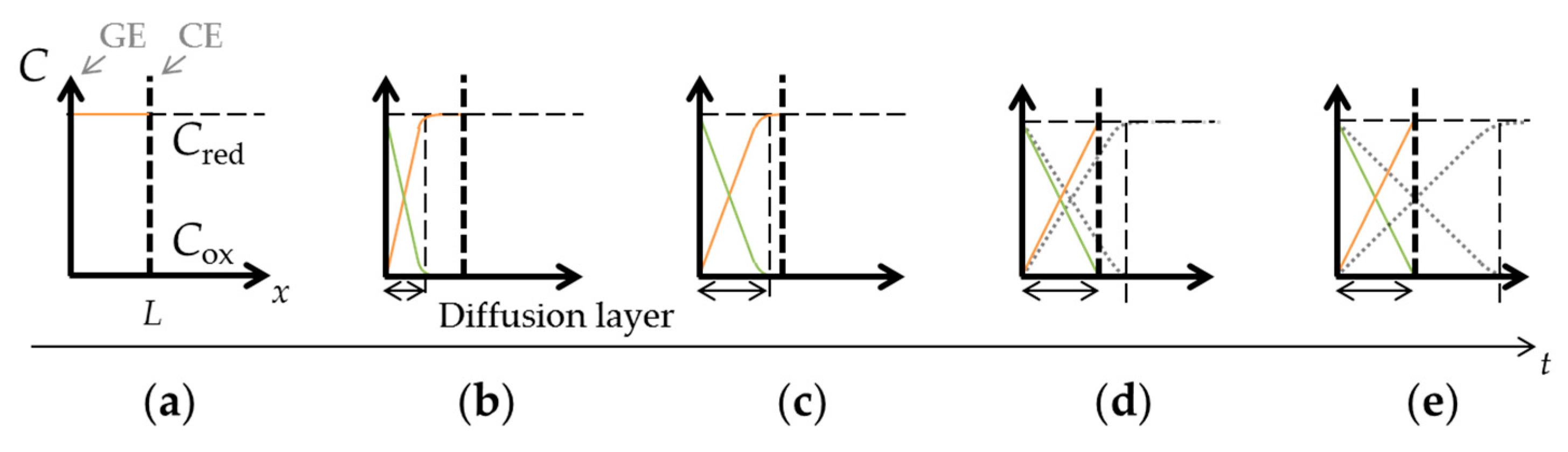
2. Theory
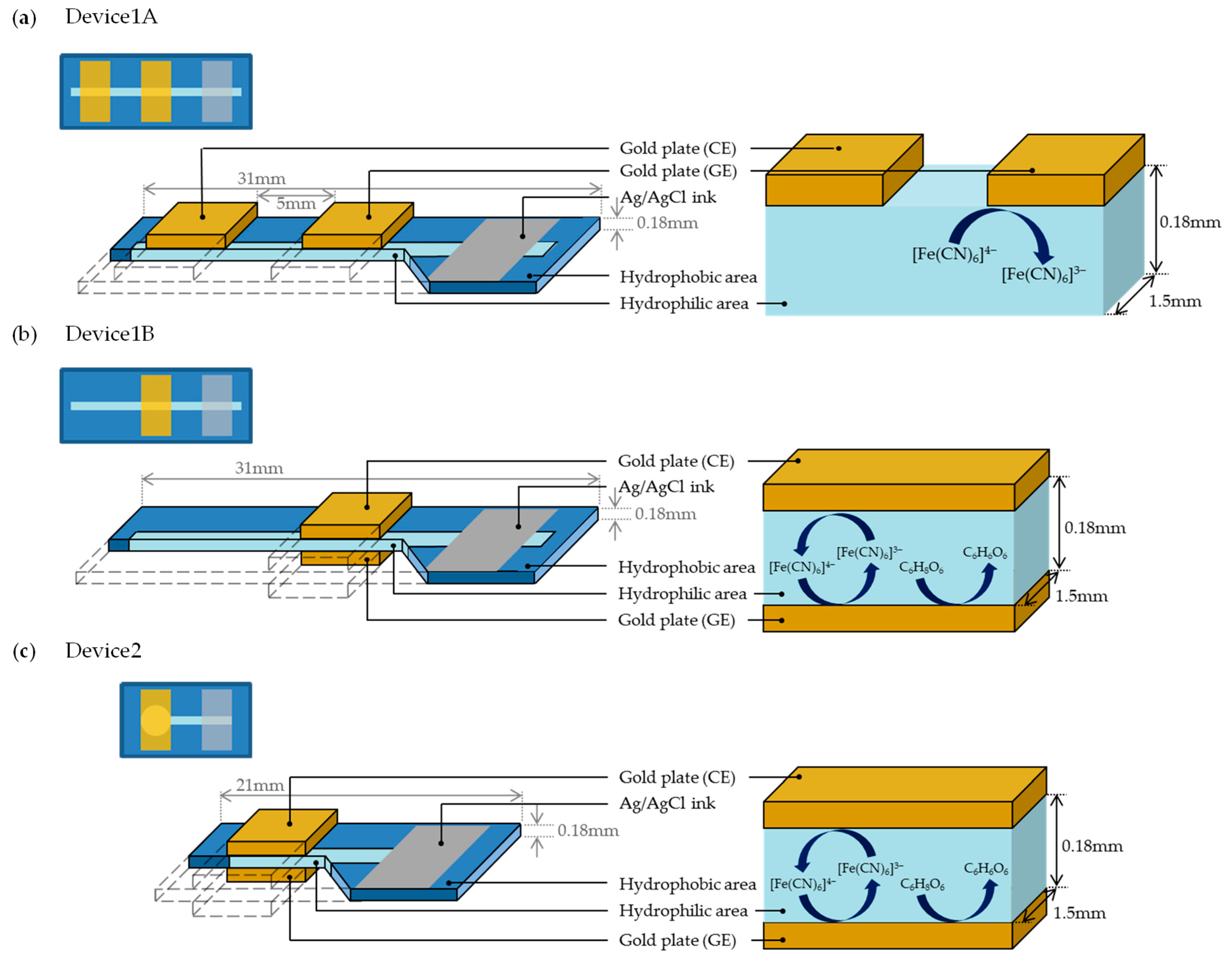
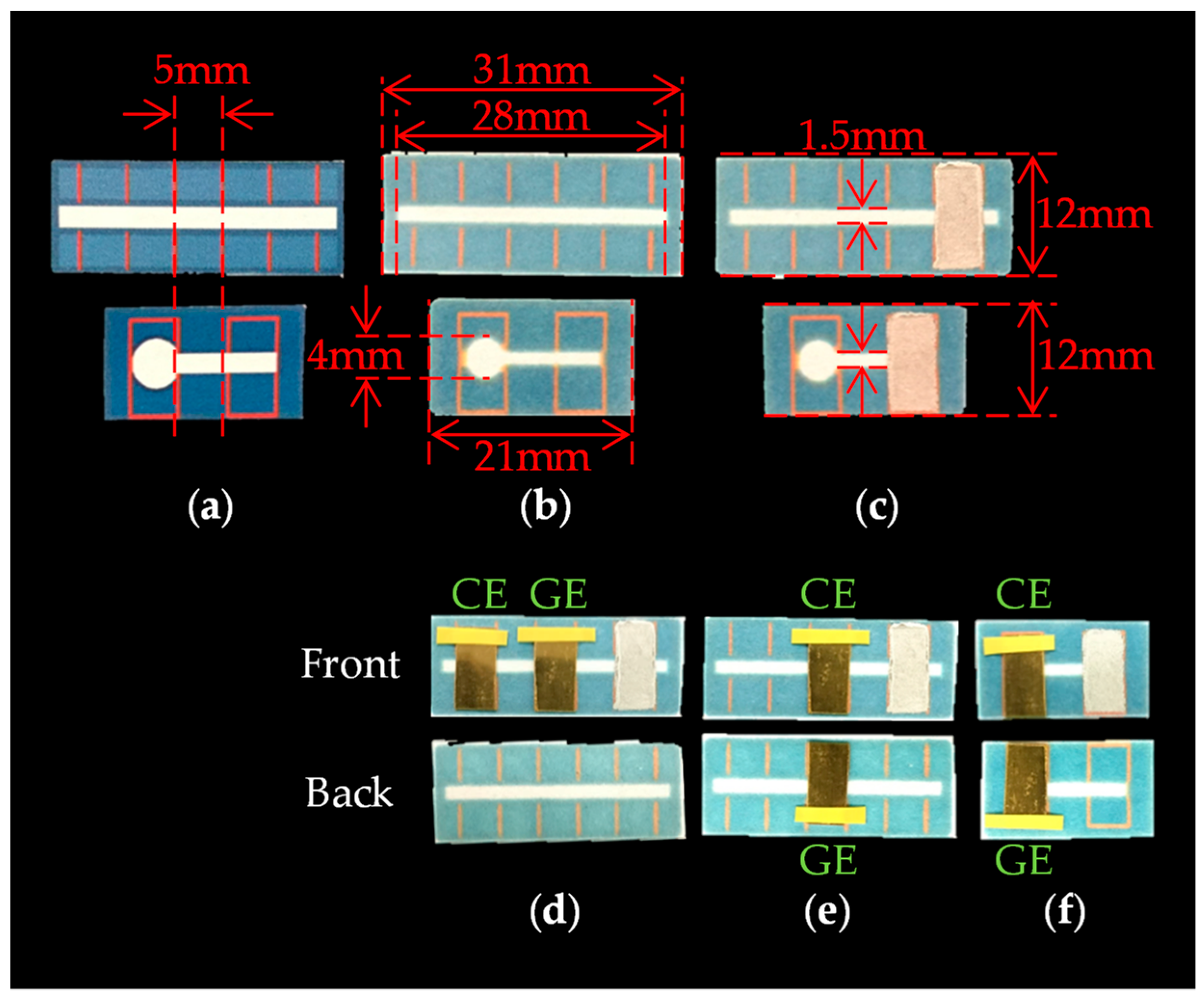
3. Experimental Method
4. Results and Discussion
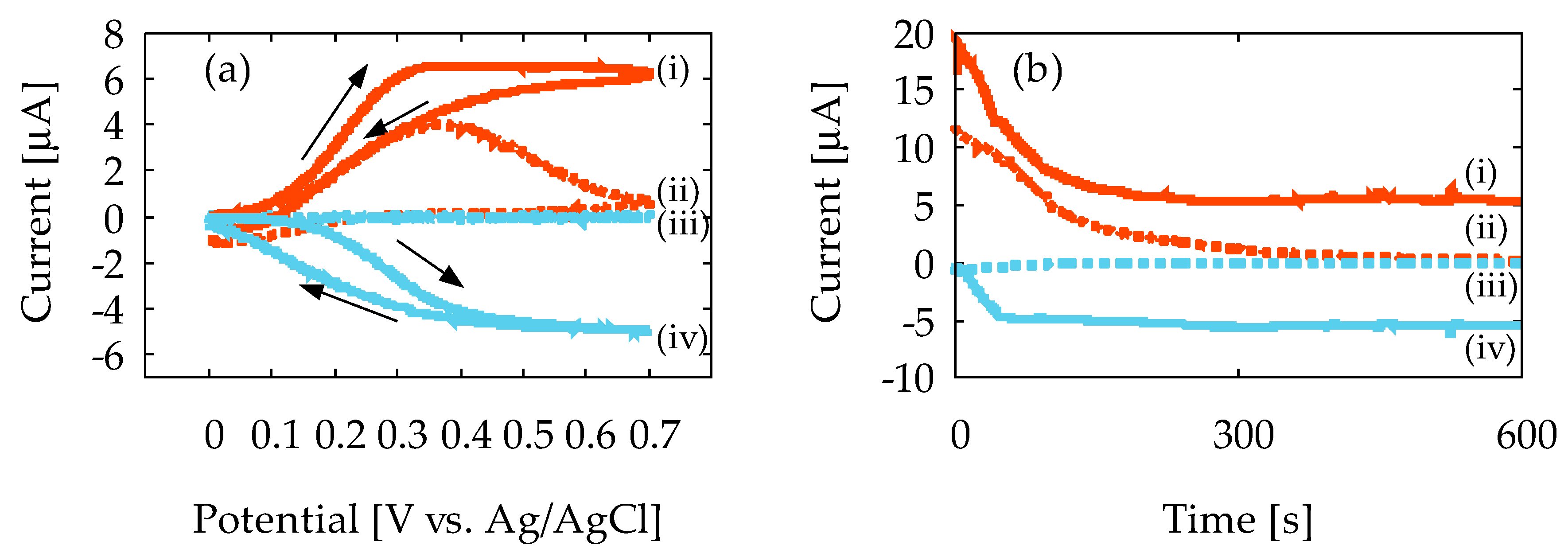
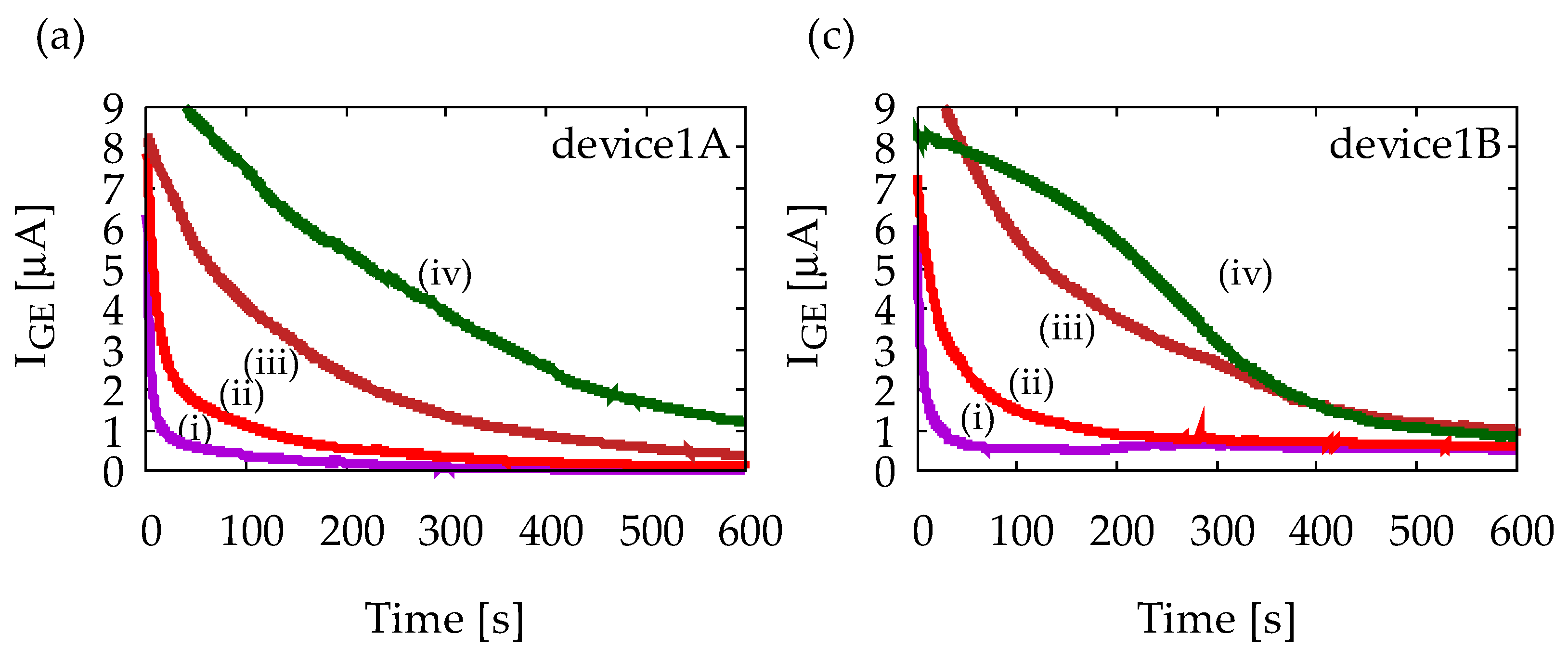
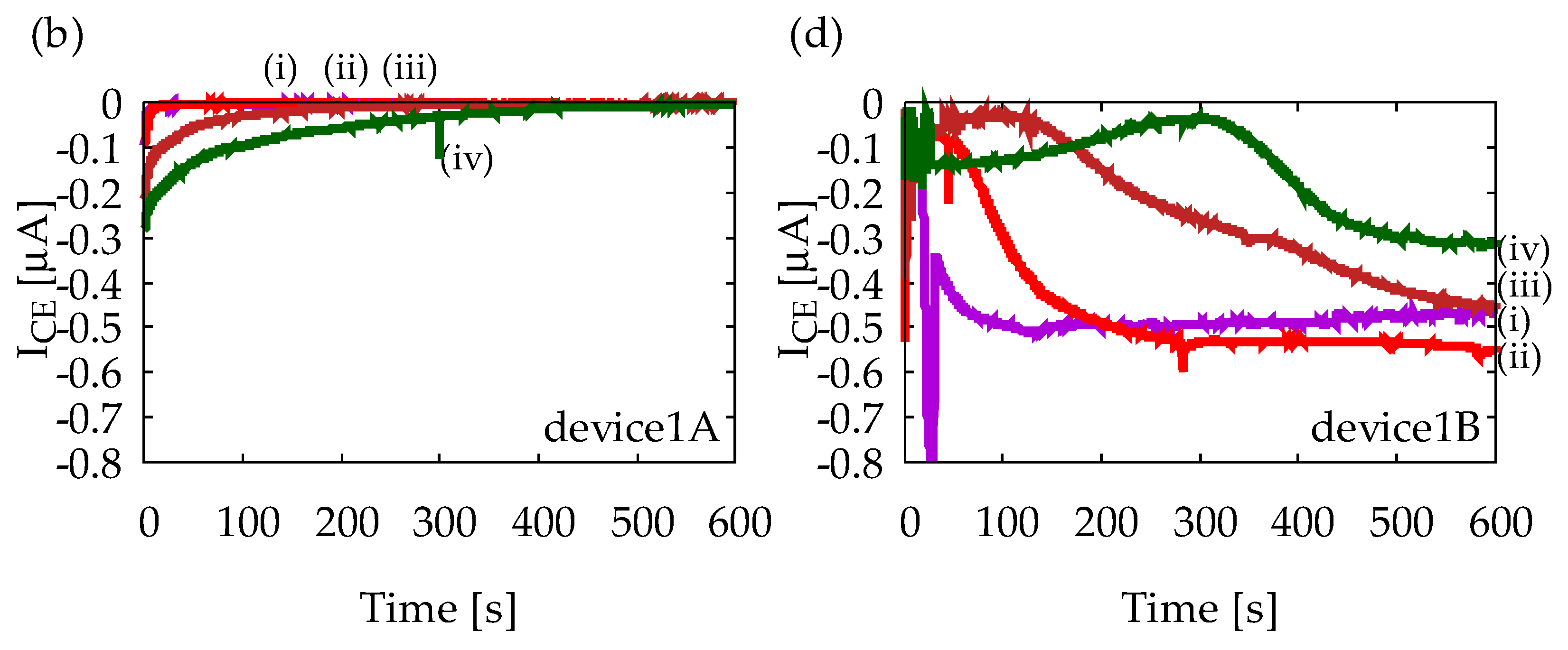
4.1. Characterization of Device1A and Device1B by Cyclic Voltammetry
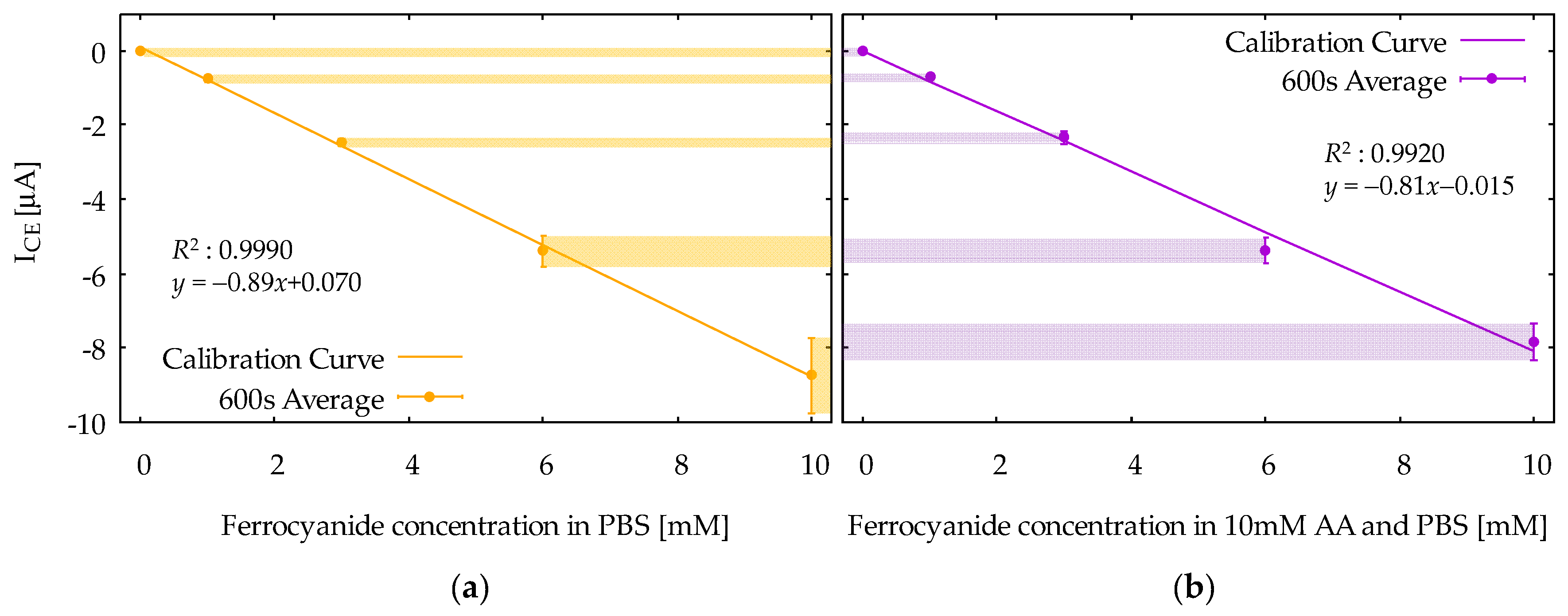
4.2. Quantitative Measurement of Ferrocyanide in Ascorbic Acid by Chronoamperometry with Device2
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A


References
- Yach, D.; Hawkes, C.; Gould, C.L.; Hofman, K.J. The Global Burden of Chronic Diseases. J. Am. Med. Assoc. 2004, 291, 2616–2622. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.; Sicree, R.; Roglic, G.; King, H.; Green, A. Global Prevalence of Diabetes. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.J.; Basu, A. Biomarkers in diabetes: Hemoglobin A1c, vascular and tissue markers. Transl. Res. 2012, 159, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [PubMed]
- Bakker, E.; Qin, Y. Electrochemical Sensors. Anal. Chem. 2006, 78, 2965–3984. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Bong, S.; Kang, Y.J.; Yang, Y.; Mahajan, R.K.; Kim, J.S.; Kim, H. Electrochemical detection of dopamine in the presence of ascorbic acid using graphene modified electrodes. Biosens. Bioelectron. 2010, 25, 2366–2369. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Hu, M.; Fritsch, I. Detection of dopamine in the presence of excess ascorbic acid at physiological concentrations through redox cycling at an unmodified microelectrode array. Anal. Bioanal. Chem. 2013, 405, 3859–3869. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Cho, C.H.; Kim, K.B.; Lee, M.H.; Kim, J.H.; Lee, S.; Cho, J.; Jung, S.; Kim, D.M.; Shim, Y.B. Interference Reduction in Glucose Detection by Redox Potential Tuning: New Glucose Meter Development. Anal. Sci. 2015, 31, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Borsook, H.; Davenport, H.W.; Jeffereys, C.E.P.; Warner, R.C. The oxidation of ascorbic acid and its reduction in vitro and in vivo. J. Biol. Chem. 1937, 117, 237–279. [Google Scholar]
- Robinson, D.L.; Hermans, A.; Speipel, A.T.; Wightman, R.M. Monitoring Rapid Chemical Communication in the Brain. Chem. Rev. 2008, 108, 2554–2584. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Rahman, M.S.; Ahmed, E.; Bakshi, P.K.; Shaikh, A.A. A Cyclic tammetric Study of the Redox Reaction of Cu(II) in Presence of Ascorbic Acid in Different pH Media. Dhaka Univ. J. Sci. 2013, 61, 161–166. [Google Scholar] [CrossRef]
- Jin, P.; Yamaguchi, A.; Asarioi, F.; Matsuo, S.; Tan, J.; Misawa, H. Glucose Sensing Based on Interdigitated Array Microelectrode. Anal. Sci. 2001, 17, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Dam, V.A.T.; Olthuis, W.; Berg, A.V.D. Redox cycling with facing interdigitated array electrodes as a method for selective detection of redox species. Analyst 2007, 132, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Mckelvey, K.; Edwards, M.A.; White, H.S. Redox Cycling in Nanogap Electrochemical Cells. The Role of Electrostatics in Determining the Cell Response. J. Phys. Chem. C 2016, 120, 17251–17260. [Google Scholar] [CrossRef]
- Niwa, O.; Morita, M.; Tabei, H. Highly Sensitive and Selective tammetric Detection of Dopamine with Vertically Separated Interdigitated Array Electrodes. Electroanalysis 1991, 3, 163–168. [Google Scholar] [CrossRef]
- Kim, S.K.; Hesketh, P.J.; Li, C.; Thomas, J.H.; Halsall, H.B.; Heineman, W.R. Fabrication of comb interdigitated electrodes array(IDA) for a microbead-based electrochemical assay system. Biosens. Bioelectron. 2004, 20, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yang, H. Sensitive and selective trypsin detection using redox cycling in the presence of l-ascorbic acid. Analyst 2014, 139, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Goluch, E.D.; Wolfrum, B.; Singh, P.S.; Zevenbergen, M.A.G.; Lemay, S. G Redox cycling in nanofluidic channels using interdigitated electrode. Anal. Bioanal. Chem. 2009, 394, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Randhahn, J.; Spilker, B.; Flehr, J.; Grabow, H.; Neudeck, A.; Jeroschewski, P.; Beikirch, H. Mathematical model of electrochemical behavior of redox cycling sensors. J. Electroanal. Chem. 2008, 613, 109–117. [Google Scholar] [CrossRef]
- Barnes, E.O.; Lewis, G.E.M.; Dale, S.E.C.; Marken, F.; Compton, R.G. Generator-collector double electrode systems: A review. Analyst 2012, 137, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Wollenberger, U.; Hintsche, R.; Scheller, F. Biosensors for analytical microsystems. Analyst 1994, 119, 1245–1249. [Google Scholar] [CrossRef]
- Ma, C.; Contento, N.M.; Gibson, L.R.; Bohn, P.W. Redox Cycling in Nanoscale-Recessed Ring-Disk Electrode Arrays for Enhanced Electrochemical Sensitivity. J. Am. Chem. Soc. 2013, 7, 5483–5490. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.J.; Marken, F. Boron-doped diamond dual-plate microtrech electrode for generator-collector chloride/chlorine sensing. Electrochem. Commun. 2014, 46, 120–123. [Google Scholar] [CrossRef]
- Hammond, J.L.; Gross, A.J.; Estrela, P.; Iniesta, J.; Green, S.J.; Winlove, C.P.; Winyard, P.G.; Benjamin, N.; Marken, F. Cysteine-Cystine Redox cycling in a gold-gold Dual-Plate Generator-Collector Microtrench Sensor. Anal. Chem. 2014, 86, 6748–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnat, M.A.; Gross, A.J.; Dale, S.E.C.; Barnes, E.O.; Compton, R.G.; Marken, F. A dual-plate ITO-ITO generator-collector microtrench sensor: Surface activation, spatial separation and suppression of irreversible oxygen and ascorbate interference. Analyst 2014, 139, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Dale, S.E.C.; Vuorema, A.; Sillanpaa, M.; Weber, J.; Wain, A.J.; Barnes, E.O.; Compton, R.G.; Marken, F. Nano-Litre Proton/Hydrogen Titration in a Dual-Plate Platinum-Platinam Generator-Collector Electrode Micro-Trench. Electrochim. Acta 2014, 125, 94–100. [Google Scholar] [CrossRef]
- Marken, F.; Mathwig, K. Nano- and micro-gap electrochemical transducer: Novel benchtop fabrication tequniques and electrical migration effects. Curr. Opin. Electrochem. 2018, 7, 15–21. [Google Scholar] [CrossRef]
- Park, S.; Park, J.H.; Hwang, S.; Kwak, J. Benchi-top fabrication and electrochemical applications of a micro-gap electrode using a microbead spacer. Electrochem. Commun. 2016, 68, 76–80. [Google Scholar] [CrossRef]
- Ben-Yoav, H.; Winkler, T.E.; Kim, E.; Chocron, S.E.; Kelly, D.L.; Payne, G.F.; Ghodssi, R. Redox cycling-based amplifying electrochemical sensor for in situ clozapine antipsychotic treatment monitoring. Electrochim. Acta 2014, 130, 497–503. [Google Scholar] [CrossRef]
- Liana, D.D.; Raguse, B.; Gooding, J.J.; Chow, E. Recent Advances in Paper-Based Sensors. Sensors 2012, 12, 11505–11526. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Phillips, S.T.; Butte, M.J.; Whitesides, G.M. Patterned Paper as a Platform for Inexpensive, Low ume, Portable Bioassays. Angew. Chem. Int. Ed. Engl. 2007, 46, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Nijhuis, C.A.; Gong, J.; Chen, X.; Kumachev, A.; Martinez, A.W.; Narovlyansky, M.; Whitesides, G.M. Electrochemical sensing in paper-based microfluidic devices. Lab Chip 2010, 10, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Santhiago, M.; Wydallis, J.B.; Kubota, L.T.; Henry, C.S. Construction and Electrochemical Characterization of Microelectrodes for Improved Sensitivity in Paper-Based Analytical Devices. Anal. Chem. 2013, 85, 5233–5239. [Google Scholar] [CrossRef] [PubMed]
- Fukayama, K.; Yamamoto, S.; Uno, S. Enhancement of Electrochemical Current by Redox Cycling Realized by Chromatography Paper-based Sensor. Telekomnika 2017, 15, 842–846. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; David, H., Elizabeth, S., Charity, R., Eugene, A., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2001; pp. 163–164, 813. ISBN 13 978-0-471-04372-0. [Google Scholar]
- Huang, T.H.; Salter, G.; Kahn, S.L.; Gind, Y.M. Redox Titration of Ferricyanide to Ferrocyanide with Ascorbic Acid: Illustrating the Nernst Equation and Beer-Lambert Law. J. Chem. Educ. 2007, 84, 1461–1463. [Google Scholar] [CrossRef]
- Dekker, M.C.J.; Bonifati, V.; Duijn, C.M.V. Parkinson’s disease: Pecing together a genetic jigsaw. Adv. Access Publ. 2013, 126, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; EisenHofer, G.; Kopin, I.J. Sources and Significance of Plasma Levels of Catechols and Their Metabolites in Humans. J. Pharmacol. Exp. Ther. 2003, 305, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Ozqan, A.; Sahin, Y. Selective and Sensitive Voltammetric Determination of Dopamine in Blood by Electrochemically Treated Pencil Graphite Electrodes. Electroanalysis 2009, 21, 2363–2370. [Google Scholar] [CrossRef]
- Niwa, O.; Morita, M.; Tabei, H. Highly Selective Electrochemical Detection of Dopamine using Interdigitated Array Electrodes Modified with Nafion/Polyester lonomer Layered Film. Electroanalysis 1994, 6, 237–243. [Google Scholar] [CrossRef]
- Sharma, D.; Lim, Y.; Lee, Y.; Shin, H. Glucose sensor based on redox-cycling between selectively modified and unmodified combs of carbon interdigitated array nanoelectrodes. Anal. Chim. Acta 2015, 889, 194–202. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement Technique | Applied Voltage (mV) | Sweep Rate (mV/s) | Measurement Time (s) | |
|---|---|---|---|---|
| GE | CE | |||
| CV | 0 to +700 | −200 | 1.0 | 1400 |
| CA | +500 | −200 | 600 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, S.; Uno, S. Redox Cycling Realized in Paper-Based Biochemical Sensor for Selective Detection of Reversible Redox Molecules Without Micro/Nano Fabrication Process. Sensors 2018, 18, 730. https://doi.org/10.3390/s18030730
Yamamoto S, Uno S. Redox Cycling Realized in Paper-Based Biochemical Sensor for Selective Detection of Reversible Redox Molecules Without Micro/Nano Fabrication Process. Sensors. 2018; 18(3):730. https://doi.org/10.3390/s18030730
Chicago/Turabian StyleYamamoto, So, and Shigeyasu Uno. 2018. "Redox Cycling Realized in Paper-Based Biochemical Sensor for Selective Detection of Reversible Redox Molecules Without Micro/Nano Fabrication Process" Sensors 18, no. 3: 730. https://doi.org/10.3390/s18030730
APA StyleYamamoto, S., & Uno, S. (2018). Redox Cycling Realized in Paper-Based Biochemical Sensor for Selective Detection of Reversible Redox Molecules Without Micro/Nano Fabrication Process. Sensors, 18(3), 730. https://doi.org/10.3390/s18030730