Sensing Properties of Oxidized Nanostructured Silicon Surface on Vaporized Molecules

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Etching Equipment
2.2. Sample Preparation
2.3. Measuring Equipment
2.4. Measurements
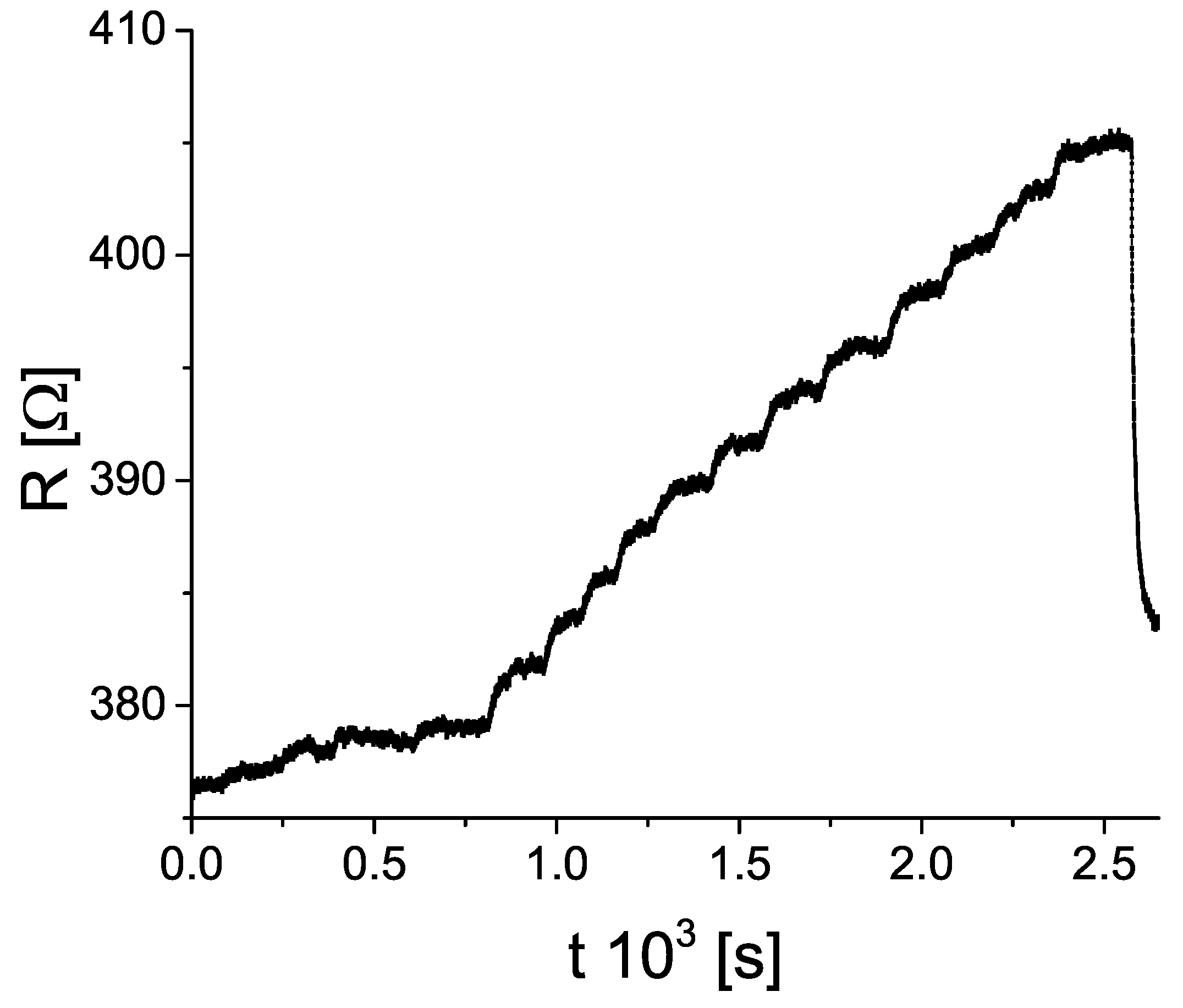
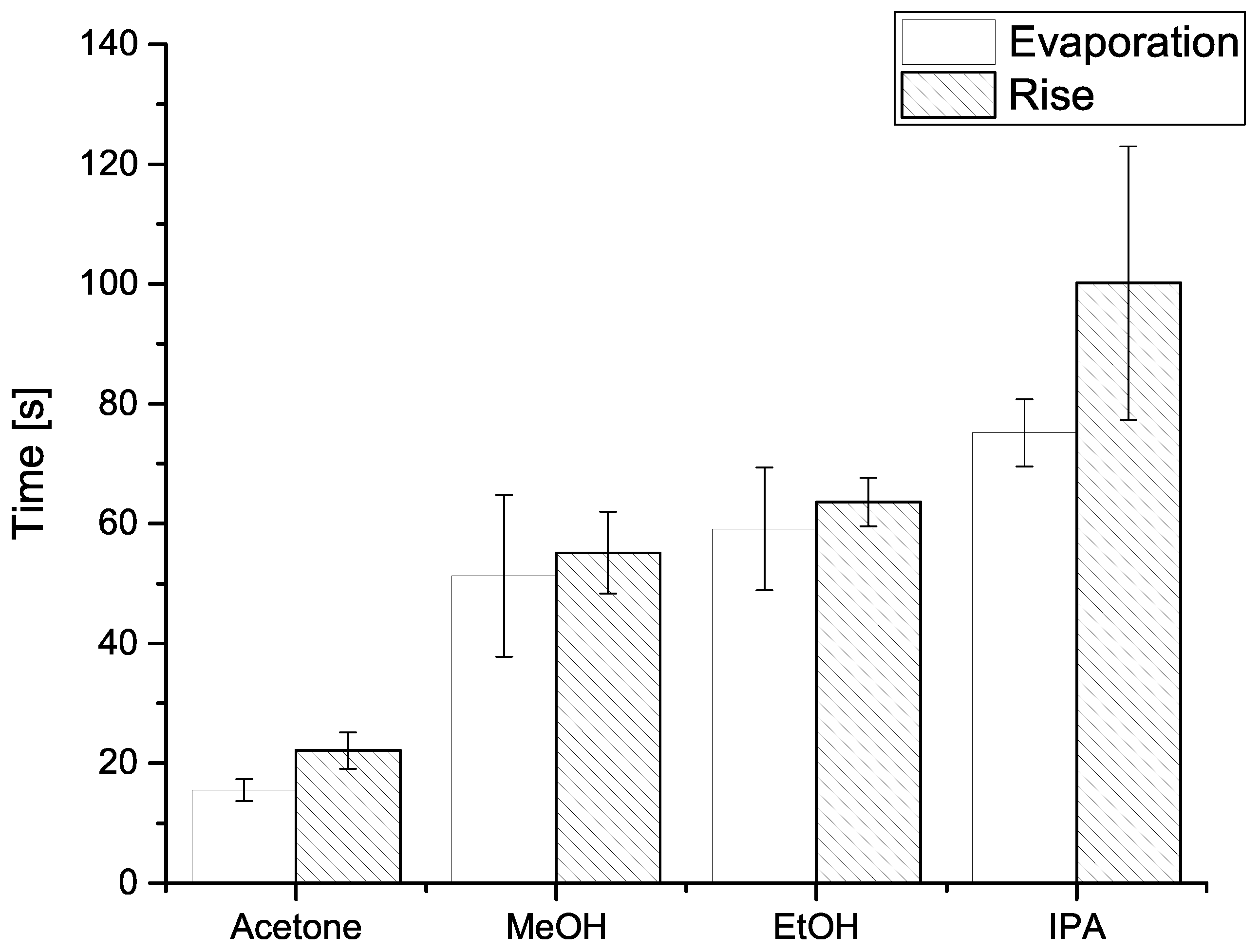
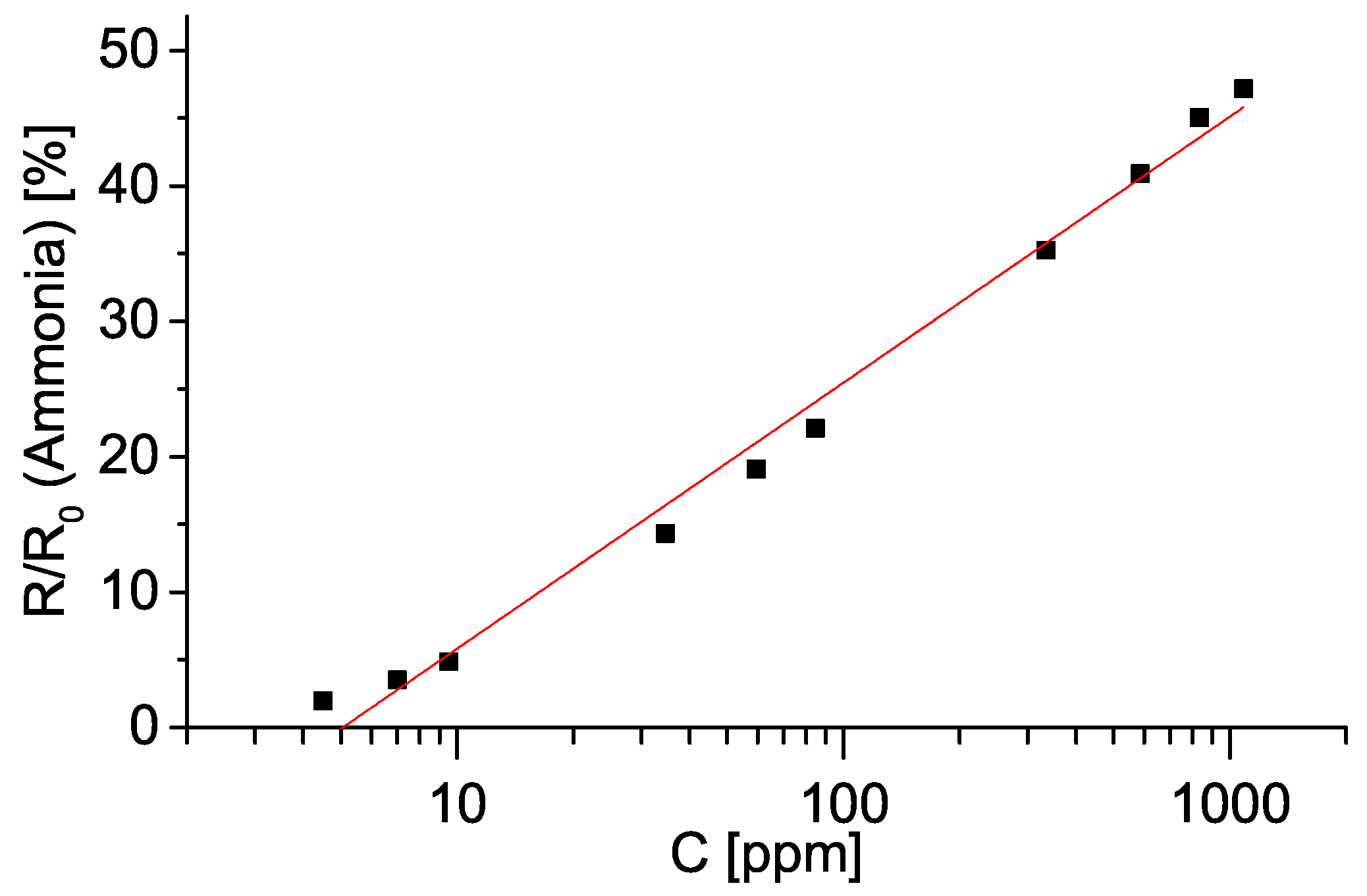
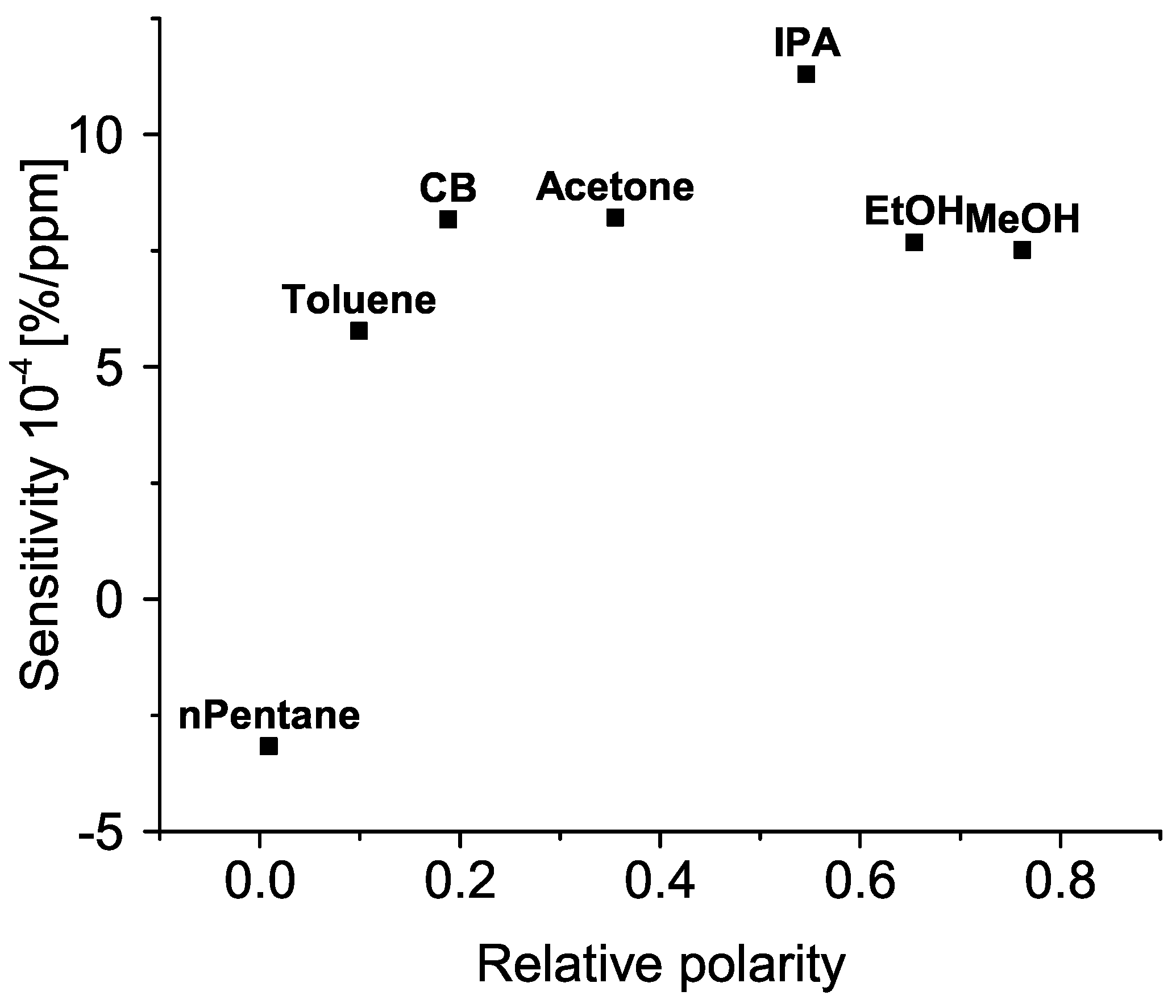
3. Results
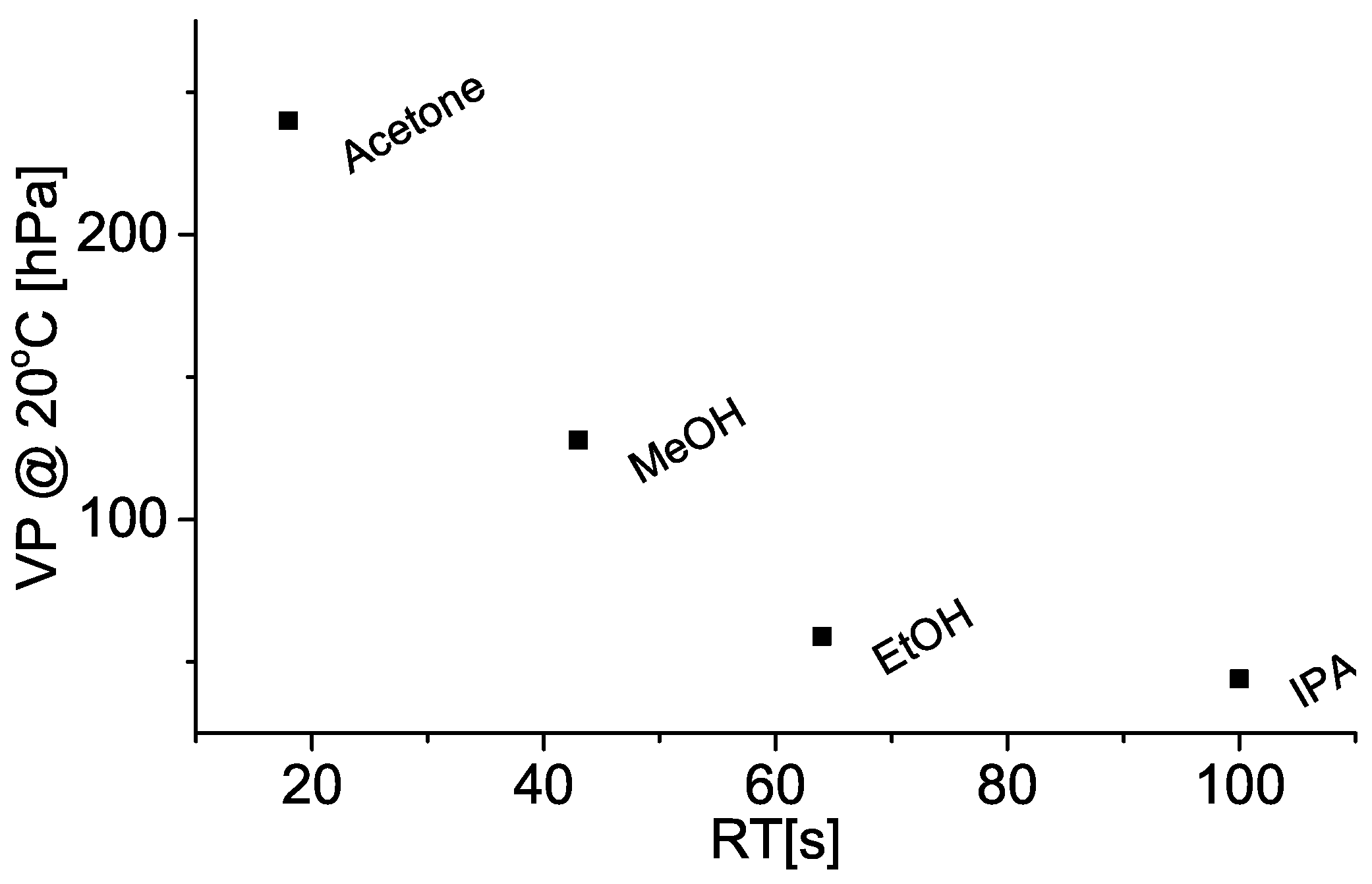
4. Discussion
4.1. Bonding Mechanism
4.2. Sensing Mechanism
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CB | Chlorobenzene |
| EDS | Energy–dispersive X–ray spectroscopy |
| EtOH | Ethanol |
| IPA | Isopropanol |
| MeOH | Methanol |
| OPS | Oxidized Porous Silicon |
| PS | Porous Silicon |
| PTFE | Polytetrafluoroethylene |
Appendix A. Measurement Parameters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isopropanol | Acetone | Chlorobenzene | Ethanol | Toluene | Methanol | Ammonia | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V [mm3] | C [ppm] | V [mm3] | C [ppm] | V [mm3] | C [ppm] | V [mm3] | C [ppm] | V [mm3] | C [ppm] | V [mm3] | C [ppm] | V [mm3] | C [ppm] |
| 79 | 1 | 1 | 83 | 60 | 1 | 0.5 | 52 | 57 | 1 | 7412 | 75 | 8 | 2 |
| 238 | 3 | 3 | 249 | 656 | 11 | 2.5 | 262 | 171 | 3 | 22,435 | 225 | 18 | 4.5 |
| 634 | 8 | 8 | 663 | 955 | 16 | 4.5 | 471 | 457 | 8 | 52,480 | 526 | 28 | 7 |
| 1426 | 18 | 18 | 1493 | 1552 | 26 | 10 | 1047 | 1028 | 18 | 82,526 | 826 | 38 | 9.5 |
| 2218 | 28 | 28 | 2322 | 20 | 2093 | 1600 | 28 | 157,641 | 1577 | 138 | 35 | ||
| 3009 | 38 | 38 | 3151 | 30 | 3140 | 2171 | 38 | 232,756 | 2329 | 238 | 60 | ||
| 3801 | 48 | 48 | 3980 | 40 | 4187 | 2742 | 48 | 307,871 | 3080 | 338 | 85 | ||
| 4593 | 58 | 58 | 4809 | 50 | 5233 | 3314 | 58 | 382,986 | 3831 | 1338 | 335 | ||
| 5385 | 68 | 68 | 5639 | 60 | 6280 | 3885 | 68 | 458,102 | 4582 | 2338 | 585 | ||
| 6177 | 78 | 78 | 6468 | 70 | 7327 | 4457 | 78 | 533,217 | 5333 | 3338 | 835 | ||
| 6969 | 88 | 88 | 7297 | 75 | 7850 | 5028 | 88 | 608,332 | 6084 | 4338 | 1085 | ||
| 7761 | 98 | 98 | 8126 | 80 | 8373 | 683,447 | 6835 | ||||||
| 8553 | 108 | 108 | 8956 | 85 | 8897 | 758562 | 7587 | ||||||
| 118 | 9785 | 90 | 9420 | 833,677 | 8338 | ||||||||
| 128 | 10,614 | 95 | 9943 | 908,792 | 9089 | ||||||||
| 100 | 10,467 | 983,907 | 9840 | ||||||||||
References
- Losada-Pérez, P.; Polat, O.; Parikh, A.N.; Seker, E.; Renner, F.U. Engineering the interface between lipid membranes and nanoporous gold: A study by quartz crystal microbalance with dissipation monitoring. Biointerphases 2018, 13, 011002. [Google Scholar] [CrossRef]
- Wannapob, R.; Thavarungkul, P.; Dawan, S.; Numnuam, A.; Limbut, W.; Kanatharana, P. A Simple and Highly Stable Porous Gold-based Electrochemical Sensor for Bisphenol A Detection. Electroanalysis 2017, 29, 472–480. [Google Scholar] [CrossRef]
- Zhou, X.; Cheng, X.; Zhu, Y.; Elzatahry, A.A.; Alghamdi, A.; Deng, Y.; Zhao, D. Ordered porous metal oxide semiconductors for gas sensing. Chin. Chem. Lett. 2018, 29, 405–416. [Google Scholar] [CrossRef]
- Dey, A. Semiconductor metal oxide gas sensors: A review. Mater. Sci. Eng. B 2018, 229, 206–217. [Google Scholar] [CrossRef]
- Aroutiounian, V. Metal oxide hydrogen, oxygen, and carbon monoxide sensors for hydrogen setups and cells. Int. J. Hydrog. Energy 2007, 32, 1145–1158. [Google Scholar] [CrossRef]
- Eranna, G.; Joshi, B.C.; Runthala, D.P.; Gupta, R.P. Oxide Materials for Development of Integrated Gas Sensors—A Comprehensive Review. Crit. Rev. Solid State Mater. Sci. 2004, 29, 111–188. [Google Scholar] [CrossRef]
- McInnes, S.; Lowe, R. Biomedical Uses of Porous Silicon. In Porous Silicon Biosensors Employing Emerging Capture Probes; Springer: Berlin/Heidelberg, Germany, 2015; Chapter 5. [Google Scholar]
- Bilyalov, R.; Stalmans, L.; Schirone, L.; Lévy-Clément, C. Use of porous silicon antireflection coating in multicrystalline silicon solar cell processing. IEEE Trans. Electron Devices 1999, 46, 2035–2040. [Google Scholar] [CrossRef]
- Ozdemir, S.; Gole, J.L. The potential of porous silicon gas sensors. Curr. Opin. Solid State Mater. Sci. 2007, 11, 92–100. [Google Scholar] [CrossRef]
- Canham, L.T. Silicon quantum wire array fabrication by electrochemical and chemical dissolution of wafers. Appl. Phys. Lett. 1990, 57, 1046–1048. [Google Scholar] [CrossRef]
- Galstyan, V.; Martirosyan, K.; Aroutiounian, V.; Arakelyan, V.; Arakelyan, A.; Soukiassian, P. Investigations of hydrogen sensors made of porous silicon. Thin Solid Films 2008, 517, 239–241. [Google Scholar] [CrossRef]
- Aroutiounian, V.; Arakelyan, V.; Galstyan, V.; Martirosyan, K.; Soukiassian, P. Hydrogen sensor made of porous silicon and covered by TiO2-x or ZnO Al thin film. IEEE Sens. J. 2009, 9, 9–12. [Google Scholar] [CrossRef]
- Baratto, C.; Faglia, G.; Comini, E.; Sberveglieri, G.; Taroni, A.; La Ferrara, V.; Quercia, L.; Di Francia, G. A novel porous silicon sensor for detection of sub-ppm NO2 concentrations. Sens. Actuators B Chem. 2001, 77, 62–66. [Google Scholar] [CrossRef]
- Pancheri, L.; Oton, C.J.; Gaburro, Z.; Soncini, G.; Pavesi, L. Very sensitive porous silicon NO2 sensor. Sens. Actuators B Chem. 2003, 89, 237–239. [Google Scholar] [CrossRef]
- Barillaro, G.; Diligenti, A.; Nannini, A.; Strambini, L.M.; Comini, E.; Sberveglieri, G. Low-concentration NO2 detection with an adsorption porous silicon FET. IEEE Sens. J. 2006, 6, 19–23. [Google Scholar] [CrossRef]
- Barillaro, G.; Strambini, L. An integrated CMOS sensing chip for NO2 detection. Sens. Actuators B Chem. 2008, 134, 585–590. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.; Lee, C. Organic vapour sensing by current response of porous silicon layer. J. Phys. D Appl. Phys. 2001, 34, 3505–3509. [Google Scholar] [CrossRef]
- Barillaro, G.; Diligenti, A.; Marola, G.; Strambini, L.M. A silicon crystalline resistor with an adsorbing porous layer as gas sensor. Sens. Actuators B Chem. 2005, 105, 278–282. [Google Scholar] [CrossRef]
- García Salgado, G.; Díaz Becerril, T.; Juárez Santiesteban, H.; Rosendo Andrés, E. Porous silicon organic vapor sensor. Optical Mater. 2006, 29, 51–55. [Google Scholar] [CrossRef]
- Kayahan, E.; Bahsi, Z.; Oral, A.; Sezer, M. Porous silicon based sensor for organic vapors. Acta Phys. Pol. A 2015, 127, 1400–1402. [Google Scholar] [CrossRef]
- Wang, W.; Gao, Y.; Tao, Q.; Liu, Y.; Zuo, J.; Ju, X.; Zhang, J. A Novel Porous Silicon Composite Sensor for Formaldehyde Detection. Chin. J. Anal. Chem. 2015, 43, 849–855. [Google Scholar] [CrossRef]
- Zangooie, S.; Bjorklund, R.; Arwin, H. Vapor sensitivity of thin porous silicon layers. Sens. Actuators B Chem. 1997, 43, 168–174. [Google Scholar] [CrossRef]
- Levitsky, I. Porous Silicon Structures as Optical Gas Sensors. Sensors 2015, 15, 19968–19991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio, E.; Urteaga, R.; Juárez, H.; Koropecki, R. Transmittance correlation of porous silicon multilayers used as a chemical sensor platform. Sens. Actuators B Chem. 2015, 213, 164–170. [Google Scholar] [CrossRef]
- Badilla, J.P.; Rojas, D.C.; López, V.; Fahlman, B.D.; Ramírez-Porras, A. Development of an organic vapor sensor based on functionalized porous silicon. Phys. Status Solidi 2011, 208, 1458–1461. [Google Scholar] [CrossRef]
- Kelly, M.T.; Bocarsly, A.B. Mechanisms of photoluminescent quenching of oxidized porous silicon applications to chemical sensing. Coord. Chem. Rev. 1998, 171, 251–259. [Google Scholar] [CrossRef]
- Tebizi-Tighilt, F.; Zane, F.; Belhaneche-Bensemra, N.; Belhousse, S.; Sam, S.; Gabouze, N. Electrochemical gas sensors based on polypyrrole-porous silicon. Appl. Surf. Sci. 2013, 269, 180–183. [Google Scholar] [CrossRef]
- Dwivedi, P.; Dhanekar, S.; Das, S.; Chandra, S. Effect of TiO2 Functionalization on Nano-Porous Silicon for Selective Alcohol Sensing at Room Temperature. J. Mater. Sci. Technol. 2017, 33, 516–522. [Google Scholar] [CrossRef]
- Bruggeman, D.A.G. Berechnung verschiedener physikalischer Konstanten von heterogenen Substanzen. I. Dielektrizitätskonstanten und Leitfähigkeiten der Mischkörper aus isotropen Substanzen. Ann. Phys. 1935, 416, 636–664. [Google Scholar] [CrossRef]
- Peng, L.; Zhai, J.; Wang, D.; Zhang, Y.; Wang, P.; Zhao, Q.; Xie, T. Size- and photoelectric characteristics-dependent formaldehyde sensitivity of ZnO irradiated with UV light. Sens. Actuators B Chem. 2010, 148, 66–73. [Google Scholar] [CrossRef]
- Das, J.; Hossain, S.; Dey, S.; Saha, H. Theoretical modeling, design, fabrication, and testing of porous-silicon-based capacitive vapor sensor. Smart Mater. Struct. Syst. 2003, 5062, 468–473. [Google Scholar] [CrossRef]
- Laminack, W.; Hardy, N.; Baker, C.; Gole, J.L. Approach to Multigas Sensing and Modeling on Nanostructure Decorated Porous Silicon Substrates. IEEE Sens. J. 2015, 15, 6491–6497. [Google Scholar] [CrossRef]
- Archer, M.; Christophersen, M.; Fauchet, P. Electrical porous silicon chemical sensor for detection of organic solvents. Sens. Actuators B Chem. 2005, 106, 347–357. [Google Scholar] [CrossRef]
- Barillaro, G.; Nannini, A.; Pieri, F. APSFET: A new, porous silicon-based gas sensing device. Sens. Actuators B Chem. 2003, 93, 263–270. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baran, N.; Gebavi, H.; Mikac, L.; Ristić, D.; Gotić, M.; Syed, K.A.; Ivanda, M. Sensing Properties of Oxidized Nanostructured Silicon Surface on Vaporized Molecules. Sensors 2019, 19, 119. https://doi.org/10.3390/s19010119
Baran N, Gebavi H, Mikac L, Ristić D, Gotić M, Syed KA, Ivanda M. Sensing Properties of Oxidized Nanostructured Silicon Surface on Vaporized Molecules. Sensors. 2019; 19(1):119. https://doi.org/10.3390/s19010119
Chicago/Turabian StyleBaran, Nikola, Hrvoje Gebavi, Lara Mikac, Davor Ristić, Marijan Gotić, Kamran Ali Syed, and Mile Ivanda. 2019. "Sensing Properties of Oxidized Nanostructured Silicon Surface on Vaporized Molecules" Sensors 19, no. 1: 119. https://doi.org/10.3390/s19010119
APA StyleBaran, N., Gebavi, H., Mikac, L., Ristić, D., Gotić, M., Syed, K. A., & Ivanda, M. (2019). Sensing Properties of Oxidized Nanostructured Silicon Surface on Vaporized Molecules. Sensors, 19(1), 119. https://doi.org/10.3390/s19010119