Comparative Study of Three Mixing Methods in Fusion Technique for Determining Major and Minor Elements Using Wavelength Dispersive X-ray Fluorescence Spectroscopy


Abstract
:
1. Introduction
2. Experimental
2.1. Instrumentation
2.2. Reagents
2.3. Reference Materials
2.4. Procedure
3. Results and Discussion
3.1. Operational Process
3.2. LLD
3.3. Accuracy and Precision
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, X.; Zou, H.; Zhang, Q.; Wei, S.; Wang, L.; Zhang, Y.; Yang, J. Application and analysis of portable X-ray fluorescence analyzer on fluorite exploration in shallow cover area. Comput. Tech. Geophys. Geochem. Explor. 2018, 40, 681–688. [Google Scholar]
- Meng, L.; Han, P.; Wang, S.; Ren, D.; Wang, J. Application progress of X-ray fluorescence spectroscopy in the detection of heavy metals in soil. Food Mach. 2017, 33, 210–213. [Google Scholar]
- Bounakha, M.; Ernbarch, K.; Zahry, F.; Biiai, E.; Kump, P. Capabilities of elemental analysis by EDXRF for geochemistry. J. Radioanal. Nucl. Chem. 2008, 275, 467–478. [Google Scholar] [CrossRef]
- Leoni, L.; Menichini, M.; Saitta, M. Determination of S, Cl and F in silicate rocks by X-Ray fluorescence analyses. X-ray Spectrom. 1982, 11, 156–158. [Google Scholar] [CrossRef]
- Ren, J.; Yan, R.; Liu, Y. Determination of trace chromium in cement by X-ray fluorescence spectrometer. Chin. J. Anal. Lab. 2015, 34, 735–739. [Google Scholar]
- Xie, L.; Wang, J.; Zhao, X.; Yang, R.; Feng, X.; Li, Y.; Luan, L.; Zhang, L.; He, R.; Liu, Y.; et al. Rapid screening of chromium-tainted gelatin capsules by wavelength dispersive X-ray fluorescence spectrometry. Chin. J. Pharm. Anal. 2013, 33, 286–288, 291. [Google Scholar]
- Smagunova, A.N.; Rozova, O.F.; Skribko, N.N. X-ray fluorescence analysis of powder products in ferrous-metallurgy-review. Ind. Lab. 1990, 56, 1062–1073. [Google Scholar]
- Pfandl, K.; Kueppers, B.; Scheiber, S.; Stockinger, G.; Holzer, J.; Pomberger, R.; Antrekowitsch, H.; Vollprecht, D. X-ray fluorescence sorting of non-ferrous metal fractions from municipal solid waste incineration bottom ash processing depending on particle surface properties. Waste Manag. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Leake, B.E.; Hendry, G.L.; Kemp, A.; Plant, A.G.; Harrey, P.K.; Wilson, J.R.; Coats, J.S.; Aucott, J.W.; Lünel, T.; Howarth, R.J. The chemical analysis of rock powders by automatic X-ray fluorescence. Chem. Geol. 1969, 5, 7–86. [Google Scholar] [CrossRef]
- Afonin, V.P. X-ray fluorescence analysis of rocks. Fresenius’ Z. Anal. Chem. 1989, 335, 54–57. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakamura, T. X-ray fluorescence analysis of rare earth elements in rocks using low dilution glass beads. Anal. Sci. 2005, 21, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrish, K.; Hutton, J.T. An accurate X-ray spectrographic method for the analysis of a wide range of geological samples. Geochim. Cosmochim. Acta 1969, 33, 431–453. [Google Scholar] [CrossRef]
- Lodha, G.S.; Sawhney, K.J.S.; Choubey, V.M. Determination of Y, Zr, Ba, La and Ce in granitic rocks using energy-dispersive X-ray fluorescence. X-ray Spectrom. 1989, 18, 225–227. [Google Scholar] [CrossRef]
- Eastell, J.; Willis, J.P. A low dilution fusion technique for the analysis of geological samples 1—Method and trace element analysis. X-ray Spectrom. 1990, 19, 3–14. [Google Scholar] [CrossRef]
- Eastell, J.; Willis, J.P. A low dilution fusion technique for the analysis of geological samples. 2—Major and minor element analysis and the use of influence/alpha coefficients. X-ray Spectrom. 1993, 22, 71–79. [Google Scholar] [CrossRef]
- Goto, A.; Tatsumi, Y. Quantitative analysis of rock samples by an X-ray fluorescence spectrometer (I). Rigaku J. 1994, 11, 40–59. [Google Scholar]
- Goto, A.; Tatsumi, Y. Quantitative analysis of rock samples by an X-ray fluorescence spectrometer (II). Rigaku J. 1996, 13, 20–38. [Google Scholar]
- Tamponi, M.; Bertoli, F.; Innocenti, F.; Leoni, L. X-ray fluorescence analysis of major elements in silicate rocks using fused glass discs. Atti Soc. Tosc. Sci. Nat. Mem. 2003, 108, 73–79. [Google Scholar]
- Ichikawa, S.; Nakamura, T. Solid Sample Preparations and Applications for X-Ray Fluorescence Analysis. In Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 1–22. [Google Scholar] [CrossRef]
- Ogasawara, M.; Mikoshiba, M.; Geshi, N.; Shimoda, G.; Ishizuka, Y. Optimization of analytical conditions for major element analysis of geological samples with XRF using glass beads. Bull. Geol. Surv. Jpn. 2018, 69, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Tani, K.; Kawabata, H.; Chang, Q.; Sato, K.; Tatsumi, Y. Quantitative analyses of silicate rock major and trace elements by X-ray fluorescence spectrometer: Evaluation of analytical precision and sample preparation. Front. Res. Earth Evol. 2005, 2, 1–8. [Google Scholar]
- Orihashi, Y.; Hirata, T. Rapid quantitative analysis of Y and REE abundances in XRF glass bead for selected GSJ reference rock standards using Nd-YAG 266 nm UV laser ablation ICP-MS. Geochem. J. 2003, 37, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Stern, W.B. On trace element analysis of geological samples by X-ray fluorescence. X-ray Spectrom. 1976, 5, 56–60. [Google Scholar] [CrossRef]
- Yamasaki, T. XRF major element analyses of silicate rocks using 1:10 dilution ratio glass bead and a synthetically extended calibration curve method. Bull. Geol. Surv. Jpn. 2014, 65, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Kamei, A. Determination of trace element abundances in GSJ reference rock samples using lithium metaborate–lithium tetraborate fused solutions and inductively coupled plasma mass spectrometry. Geosci. Rept. Shimane Univ. 2016, 34, 41–49. [Google Scholar]
- Krusberski, N. Exploring Potential Errors in XRF Analysis; The Southern African Institute of Mining and Metallurgy Analytical Challenges in Metallurgy: Johannesburg, South Africa, 2006; pp. 1–8. [Google Scholar]
- GmbH, R. Representative Sample Preparation for XRF Analysis. AZoM. Available online: https://www.azom.com/article.aspx?ArticleID=16242 (accessed on 8 September 2020).
- Muller, R. Spectrochmical Analysis by X-ray Fluorescence; Plenum Press: New York, NY, USA, 1972. [Google Scholar]
- Anzelmo, J.; Bouchard, M.; Provencher, M.E. Atomic Perspectives X-ray Fluorescence Spectroscopy, Part II: Sample Preparation. Spectrosc. Springf. Eugene Duluth 2014, 29, 12. [Google Scholar]
- Lupton, D.F.; Merker, J.; Schölz, F. The Correct Use of Platinum in the XRF Laboratory. X-ray Spectrom. 1997, 26, 132–140. [Google Scholar] [CrossRef]
- Shrivastava, A.; Gupta, V. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. 1), S49–S52. [Google Scholar]
- Gazulla, M.F.; Rodrigo, M.; Vicente, S.; Orduña, M. Methodology for the determination of minor and trace elements in petroleum cokes by wavelength-dispersive X-ray fluorescence (WD-XRF). X-ray Spectrom. 2010, 39, 321–327. [Google Scholar] [CrossRef]
- Han, X. Determination of Palladium, Platinum and Gold by X-ray Fluorescence Spectrometry with Resin Separation; Yantai University: Yantai, China, 2013. [Google Scholar]
- Whitty-Léveillé, L.; Turgeon, K.; Bazin, C.; Larivière, D. A comparative study of sample dissolution techniques and plasma-based instruments for the precise and accurate quantification of REEs in mineral matrices. Anal. Chim. Acta 2017, 961, 33–41. [Google Scholar] [CrossRef]
- Anscombe, F.J.; Tukey, J.W. The Examination and Analysis of Residuals. Technometrics 1963, 5, 141–160. [Google Scholar] [CrossRef]
- Stix, J.; Gorton, M.P.; Fontaine, E. Major and trace element analysis of fifteen Japanese igneous reference rocks by XRFS and INAA. Geostand. Newsl. 1996, 20, 87–94. [Google Scholar] [CrossRef]
- Herbrich, A.; Hauff, F.; Hoernle, K.; Werner, R.; Garbe-Schönberg, D.; White, S. A 1.5 Ma record of plume-ridge interaction at the Western Galápagos Spreading Center (91°40′–92°00′ W). Geochim. Cosmochim. Acta 2016, 185, 141–159. [Google Scholar] [CrossRef] [Green Version]
- Golowin, R.; Portnyagin, M.; Hoernle, K.; Hauff, F.; Werner, R.; Garbe-Schönberg, D. Geochemistry of deep Manihiki Plateau crust: Implications for compositional diversity of large igneous provinces in the Western Pacific and their genetic link. Chem. Geol. 2018, 493, 553–566. [Google Scholar] [CrossRef]
- Yu, Z.; Norman, M.D.; Robinson, P. Major and trace element analysis of silicate rocks by XRF and laser ablation ICP-MS using lithium borate fused glasses: Matrix Effects, Instrument Response and Results for International Reference Materials. Geostand. Newsl. 2003, 27, 67–89. [Google Scholar] [CrossRef]
- Freund, S.; Haase, K.M.; Keith, M.; Beier, C.; Garbe-Schönberg, D. Constraints on the formation of geochemically variable plagiogranite intrusions in the Troodos Ophiolite, Cyprus. Contrib. Mineral. Petrol. 2014, 167, 978. [Google Scholar] [CrossRef]
- Raczek, I.; Stoll, B.; Hofmann, A.W.; Peter Jochum, K. High-precision trace element data for the USGS reference materials BCR-1, BCR-2, BHVO-1, BHVO-2, AGV-1, AGV-2, DTS-1, DTS-2, GSP-1 and GSP-2 by ID-TIMS and MIC-SSMS. Geostand. Newsl. 2001, 25, 77–86. [Google Scholar] [CrossRef]
- McHenry, L.J. Element mobility during zeolitic and argillic alteration of volcanic ash in a closed-basin lacustrine environment: Case study Olduvai Gorge, Tanzania. Chem. Geol. 2009, 265, 540–552. [Google Scholar] [CrossRef]
- Zhang, G.; Smith-Duque, C.; Tang, S.; Li, H.; Zarikian, C.; D’Hondt, S.; Inagaki, F. Geochemistry of basalts from IODP site U1365: Implications for magmatism and mantle source signatures of the mid-Cretaceous Osbourn Trough. Lithos 2012, 144–145, 73–87. [Google Scholar] [CrossRef]
- Zhang, G.-L.; Chen, L.-H.; Li, S.-Z. Mantle dynamics and generation of a geochemical mantle boundary along the East Pacific Rise–Pacific/Antarctic ridge. Earth Planet. Sci. Lett. 2013, 383, 153–163. [Google Scholar] [CrossRef]
- Taneja, R.; Rushmer, T.; Blichert-Toft, J.; Turner, S.; O’Neill, C. Mantle heterogeneities beneath the Northeast Indian Ocean as sampled by intra-plate volcanism at Christmas Island. Lithos 2016, 262, 561–575. [Google Scholar] [CrossRef]
- Alloway, B.V.; Pearce, N.J.G.; Villarosa, G.; Outes, V.; Moreno, P.I. Multiple melt bodies fed the AD 2011 eruption of Puyehue-Cordón Caulle, Chile. Sci. Rep. 2015, 5, 17589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Vieten, K.; Romer, R.L.; Mezger, K.; Hoernes, S.; Satir, M. Petrogenesis of Tertiary Alkaline Magmas in the Siebengebirge, Germany. J. Petrol. 2012, 53, 2381–2409. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Prytulak, J.; Xu, J.; Wei, W.; Hammond, J.O.S.; Zhao, B. The cause and source of melting for the most recent volcanism in Tibet: A combined geochemical and geophysical perspective. Lithos 2017, 288–289, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Guo, F.; Yan Wang, C.; Li, C. Crustal recycling processes in generating the early Cretaceous Fangcheng basalts, North China Craton: New constraints from mineral chemistry, oxygen isotopes of olivine and whole-rock geochemistry. Lithos 2013, 170–171, 1–16. [Google Scholar] [CrossRef]
- Cheng, C.; Zheng, H.; Kapsiotis, A.; Liu, W.; Lenaz, D.; Velicogna, M.; Zhong, L.; Huang, Q.; Yuan, Y.; Xia, B. Geochemistry and geochronology of dolerite dykes from the Daba and Dongbo peridotite massifs, SW Tibet: Insights into the style of mantle melting at the onset of Neo-Tethyan subduction. Lithos 2018, 322, 281–295. [Google Scholar] [CrossRef]
- Phillips, E.H.; Sims, K.W.W.; Sherrod, D.R.; Salters, V.J.M.; Blusztajn, J.; Dulai, H. Isotopic constraints on the genesis and evolution of basanitic lavas at Haleakala, Island of Maui, Hawaii. Geochim. Cosmochim. Acta 2016, 195, 201–225. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Lassiter, J.C.; Ramirez, G. Origin of temporal compositional trends in monogenetic vent eruptions: Insights from the crystal cargo in the Papoose Canyon sequence, Big Pine Volcanic Field, CA. Earth Planet. Sci. Lett. 2017, 457, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-Q.; Xu, Y.-G.; Tian, W.; Zhong, Y.-T.; Mundil, R.; Li, X.-H.; Yang, Y.-H.; Luo, Z.-Y.; Shang-Guan, S.-M. Origin of two types of rhyolites in the Tarim Large Igneous Province: Consequences of incubation and melting of a mantle plume. Lithos 2014, 204, 59–72. [Google Scholar] [CrossRef]
- He, B.; Zhong, Y.-T.; Xu, Y.-G.; Li, X.-H. Triggers of Permo-Triassic boundary mass extinction in South China: The Siberian Traps or Paleo-Tethys ignimbrite flare-up? Lithos 2014, 204, 258–267. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Si | Ti | Al | Fe | Mn | Mg | Ca | Na | K | P | Cr | Cu | Ba | Ni | Sr | V | Zr | Zn | |
| Line | Kα | Kα | Kα | Kα | Kα | Kα | Kα | Kα | Kα | Kα | Kα | Kα | Lα | Kα | Kα | Kα | Kα | Kα | |
| Crystal | PE 002 | LiF 200 | PE 002 | LiF 200 | LiF 200 | PX1 | LiF 200 | PX1 | LiF 200 | Ge 111 | LiF 200 | LiF 200 | LiF 200 | LiF 200 | LiF 200 | LiF 200 | LiF 200 | LiF 200 | |
| Detector | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Flow | Scint. | Flow | Scint. | Scint. | |
| Voltage/kV | 30 | 40 | 30 | 60 | 60 | 30 | 30 | 30 | 30 | 30 | 40 | 60 | 40 | 60 | 60 | 40 | 60 | 60 | |
| Intensity/mA | 120 | 90 | 120 | 60 | 60 | 120 | 120 | 120 | 120 | 120 | 90 | 60 | 90 | 60 | 60 | 90 | 60 | 60 | |
| Collimator/μm | 150 | 300 | 300 | 300 | 300 | 300 | 150 | 700 | 300 | 300 | 300 | 150 | 300 | 150 | 150 | 300 | 150 | 150 | |
| Counting time/s | 60 | 30 | 40 | 26 | 36 | 40 | 40 | 40 | 30 | 30 | 40 | 40 | 60 | 40 | 40 | 40 | 40 | 40 | |
| The Grinding Method | Peak Angle/2θ | 109.0896 | 86.1196 | 144.7838 | 57.5056 | 62.9604 | 23.2312 | 113.0678 | 28.0754 | 136.6656 | 141.1432 | 69.3396 | 45.0206 | 87.1566 | 48.6566 | 25.1452 | 76.9228 | 22.5018 | 41.7962 |
| Bg1 | 2.2312 | 1.7800 | −1.6878 | 0.9264 | 0.9414 | 2.3962 | −0.9160 | −2.2264 | 2.6668 | −1.7890 | 0.9340 | 0.8988 | 0.9548 | 0.6472 | −0.5904 | −0.7804 | −0.8338 | 0.7076 | |
| Bg2 | 3.1288 | 1.7958 | 2.3114 | 2.5214 | 0.9642 | ||||||||||||||
| PHD | 33–67 | 38–62 | 32–69 | 37–63 | 33–55 | 30–72 | 35–65 | 30–70 | 35–65 | 30–56 | 38–62 | 36–53 | 33–53 | 35–53 | 30–67 | 33–53 | 30–69 | 29–67 | |
| The Stirring Rod Method | Peak Angle/2θ | 109.0896 | 86.1188 | 144.7788 | 57.5038 | 62.9588 | 23.2312 | 113.0678 | 28.0660 | 136.6650 | 141.1414 | 69.3388 | 45.0214 | 87.1556 | 48.6578 | 25.1452 | 76.9202 | 22.5038 | 41.7962 |
| Bg1 | 2.2642 | −0.9434 | −1.1734 | 0.9528 | 1.8912 | 2.3820 | −0.9674 | −1.9244 | 2.4010 | −1.7264 | 1.6960 | 1.6544 | 1.4378 | 0.8044 | −0.5490 | 1.5670 | −0.7000 | 0.8320 | |
| Bg2 | 3.1132 | 1.7726 | 2.4482 | 2.5190 | 0.7962 | ||||||||||||||
| PHD | 31–72 | 36–63 | 28–72 | 34–66 | 33–53 | 29–75 | 32–70 | 23–66 | 31–69 | 26–58 | 37–62 | 35–52 | 33–53 | 34–53 | 22–78 | 32–53 | 24–78 | 25–70 | |
| The Shaker Cup Method | Peak Angle/2θ | 109.0896 | 86.1226 | 144.7788 | 57.5056 | 63.0042 | 23.2312 | 113.0678 | 28.0708 | 136.6650 | 141.1414 | 69.3388 | 45.0214 | 87.1556 | 48.6566 | 25.1452 | 76.9258 | 22.5084 | 41.7962 |
| Bg1 | 2.3066 | 1.4588 | −1.1734 | 0.9434 | 1.7556 | 2.2216 | −1.0112 | −2.0756 | 2.6322 | −1.6698 | 1.6750 | 1.4692 | 1.4678 | 0.7302 | 0.6208 | 1.7620 | −0.5896 | 0.6358 | |
| Bg2 | 3.1338 | 1.9038 | 2.3678 | 2.7170 | 0.8426 | ||||||||||||||
| PHD | 30–71 | 35–65 | 22–78 | 36–64 | 34–52 | 29–75 | 32–69 | 24–58 | 31–71 | 26–60 | 37–63 | 36–52 | 33–53 | 34–52 | 22–78 | 33–53 | 29–69 | 26–72 | |
| Number | Elements | Range of Standard Sample Composition | LLD (μg g−1) | ||
|---|---|---|---|---|---|
| Major | (m/m% a) | GR | ST | SH | |
| 1 | SiO2 | 0.62–90.36 | 157.94 | 147.90 | 148.11 |
| 2 | TiO2 | 0.004–7.69 | 25.73 | 29.34 | 30.55 |
| 3 | Al2O3 | 0.1–59.20 | 271.70 | 161.83 | 159.69 |
| 4 | TFe2O3 b | 0.075–25.65 | 17.02 | 16.28 | 15.72 |
| 5 | MnO | 0.001–0.43 | 11.49 | 11.09 | 10.85 |
| 6 | MgO | 0.006–49.40 | 54.70 | 85.41 | 85.68 |
| 7 | CaO | 0.04–51.10 | 35.89 | 49.68 | 49.95 |
| 8 | Na2O | 0.008–10.59 | 79.77 | 77.23 | 62.53 |
| 9 | K2O | 0.003–12.81 | 13.82 | 20.37 | 19.17 |
| 10 | P2O5 | 0.002–6.06 | 17.20 | 16.67 | 14.48 |
| Minor | (μg g−1) | ||||
| 11 | Cr | 1.6–15500 | 11.13 | 11.19 | 11.32 |
| 12 | Cu | 0.82–1230 | 7.73 | 7.13 | 7.22 |
| 13 | Ba | 6.4–4000 | 53.28 | 52.67 | 52.28 |
| 14 | Ni | 0.9–3780 | 5.63 | 5.52 | 5.36 |
| 15 | Sr | 2.3–12000 | 3.82 | 4.04 | 4.05 |
| 16 | V | 0.0022–768 | 12.88 | 13.16 | 13.51 |
| 17 | Zr | 0.7–1540 | 2.06 | 2.15 | 1.72 |
| 18 | Zn | 3.5–1300 | 4.44 | 5.74 | 5.78 |
| Major Oxides | Overall Correlation Coefficient | ||
|---|---|---|---|
| The Grinding (GR) Method | The Stirring Rod (ST) Method | The Shaker Cup (SH) Method | |
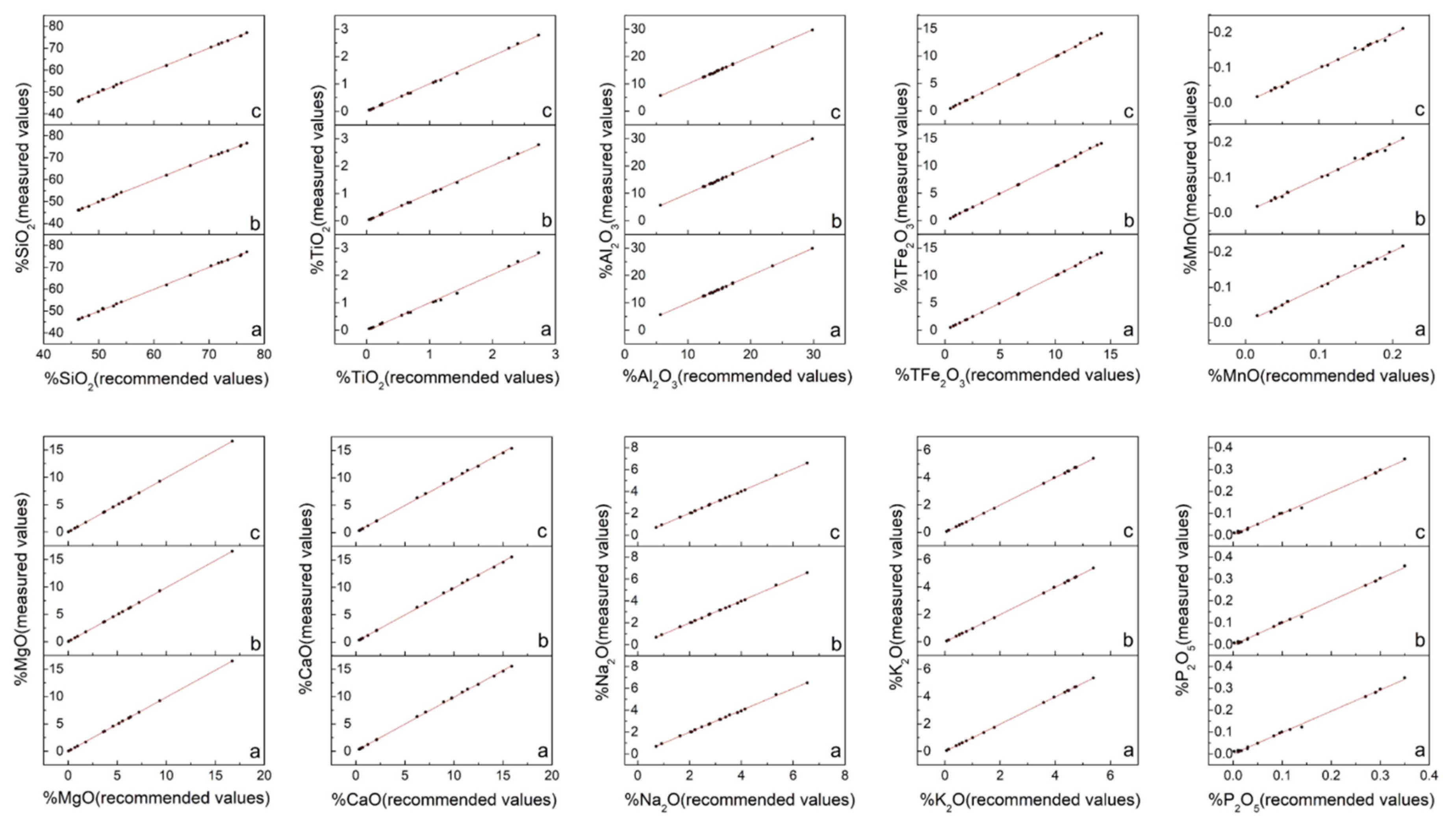
| SiO2 | 0.99958 | 0.99972 | 0.99980 |
| TiO2 | 0.99710 | 0.99893 | 0.99939 |
| Al2O3 | 0.99941 | 0.99930 | 0.99942 |
| TFe2O3 | 0.99987 | 0.99983 | 0.99983 |
| MnO | 0.99561 | 0.99649 | 0.99664 |
| MgO | 0.99995 | 0.99998 | 0.99999 |
| CaO | 0.99967 | 0.99960 | 0.99954 |
| Na2O | 0.99929 | 0.99951 | 0.99951 |
| K2O | 0.99994 | 0.99992 | 0.99990 |
| P2O5 | 0.99813 | 0.99856 | 0.99828 |
| Analytical Line | Energy/keV | Analytical Depth/μm | Analytical Line | Energy/keV | Analytical Depth/μm |
|---|---|---|---|---|---|
| Si Kα | 1.74 | 23 | P Kα | 2.01 | 21 |
| Ti Kα | 4.51 | 281 | Cr Kα | 5.41 | 530 |
| Al Kα | 1.49 | 10 | Cu Kα | 8.05 | 1080 |
| Fe Kα | 6.40 | 560 | Ba Lα | 4.47 | 272 |
| Mn Kα | 5.90 | 461 | Ni Kα | 7.48 | 916 |
| Mg Kα | 1.25 | 4 | Sr Kα | 14.17 | 3831 |
| Ca Kα | 3.69 | 158 | V Kα | 4.95 | 430 |
| Na Kα | 1.04 | 3 | Zr Kα | 15.78 | 4875 |
| K Kα | 3.31 | 95 | Zn Kα | 8.64 | 1279 |
| Elements | JST;i a | n b | JSA;i c | ||||
|---|---|---|---|---|---|---|---|
| GR | ST | SH | GR | ST | SH | ||
| Major | |||||||
| SiO2 | 5.54 | 5.47 | 4.28 | 17 | 0.33 | 0.32 | 0.25 |
| TiO2 | 16.96 | 7.2 | 4.56 | 16 | 1.06 | 0.45 | 0.29 |
| Al2O3 | 5.44 | 6.39 | 3.27 | 17 | 0.32 | 0.38 | 0.19 |
| TFe2O3 | 2.03 | 16.06 | 11.6 | 17 | 0.12 | 0.94 | 0.68 |
| MnO | 30.57 | 5.62 | 5.97 | 17 | 1.80 | 0.33 | 0.35 |
| MgO | 9.62 | 8.1 | 5.3 | 16 | 0.60 | 0.51 | 0.33 |
| CaO | 16.05 | 23.96 | 21.59 | 17 | 0.94 | 1.41 | 1.27 |
| Na2O | 1.33 | 1.46 | 2.53 | 17 | 0.08 | 0.09 | 0.15 |
| K2O | 2.91 | 7.32 | 2.91 | 17 | 0.17 | 0.43 | 0.17 |
| P2O5 | 8.31 | 11.43 | 4.25 | 16 | 0.52 | 0.71 | 0.27 |
| Minor | |||||||
| Cr | 9.65 | 3.71 | 4 | 12 | 0.80 | 0.31 | 0.33 |
| Cu | 87.84 | 22.48 | 24.1 | 9 | 9.76 | 2.50 | 2.68 |
| Ba | 2.57 | 5.24 | 7.71 | 12 | 0.21 | 0.44 | 0.64 |
| Ni | 11.35 | 7.72 | 5.57 | 10 | 1.14 | 0.77 | 0.56 |
| Sr | 55.94 | 40.44 | 44.52 | 12 | 4.66 | 3.37 | 3.71 |
| V | 7.6 | 3.61 | 4.96 | 11 | 0.69 | 0.33 | 0.45 |
| Zr | 14 | 14.83 | 91.66 | 17 | 0.82 | 0.87 | 5.39 |
| Zn | 240.05 | 44.41 | 45.96 | 16 | 15.00 | 2.78 | 2.87 |
| JS;major d | 5.94 | 5.57 | 3.95 | ||||
| JS;minor e | 33.09 | 11.36 | 16.64 | ||||
| JS f | 39.03 | 16.93 | 20.59 | ||||
| JS;without Zr g | 38.21 | 16.06 | 15.20 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.-P.; Xue, D.-S.; Liu, Y.-H.; Wan, B.; Guo, Q.; Guo, J.-J. Comparative Study of Three Mixing Methods in Fusion Technique for Determining Major and Minor Elements Using Wavelength Dispersive X-ray Fluorescence Spectroscopy. Sensors 2020, 20, 5325. https://doi.org/10.3390/s20185325
Zhang D-P, Xue D-S, Liu Y-H, Wan B, Guo Q, Guo J-J. Comparative Study of Three Mixing Methods in Fusion Technique for Determining Major and Minor Elements Using Wavelength Dispersive X-ray Fluorescence Spectroscopy. Sensors. 2020; 20(18):5325. https://doi.org/10.3390/s20185325
Chicago/Turabian StyleZhang, Dan-Ping, Ding-Shuai Xue, Yan-Hong Liu, Bo Wan, Qian Guo, and Ju-Jie Guo. 2020. "Comparative Study of Three Mixing Methods in Fusion Technique for Determining Major and Minor Elements Using Wavelength Dispersive X-ray Fluorescence Spectroscopy" Sensors 20, no. 18: 5325. https://doi.org/10.3390/s20185325
APA StyleZhang, D. -P., Xue, D. -S., Liu, Y. -H., Wan, B., Guo, Q., & Guo, J. -J. (2020). Comparative Study of Three Mixing Methods in Fusion Technique for Determining Major and Minor Elements Using Wavelength Dispersive X-ray Fluorescence Spectroscopy. Sensors, 20(18), 5325. https://doi.org/10.3390/s20185325