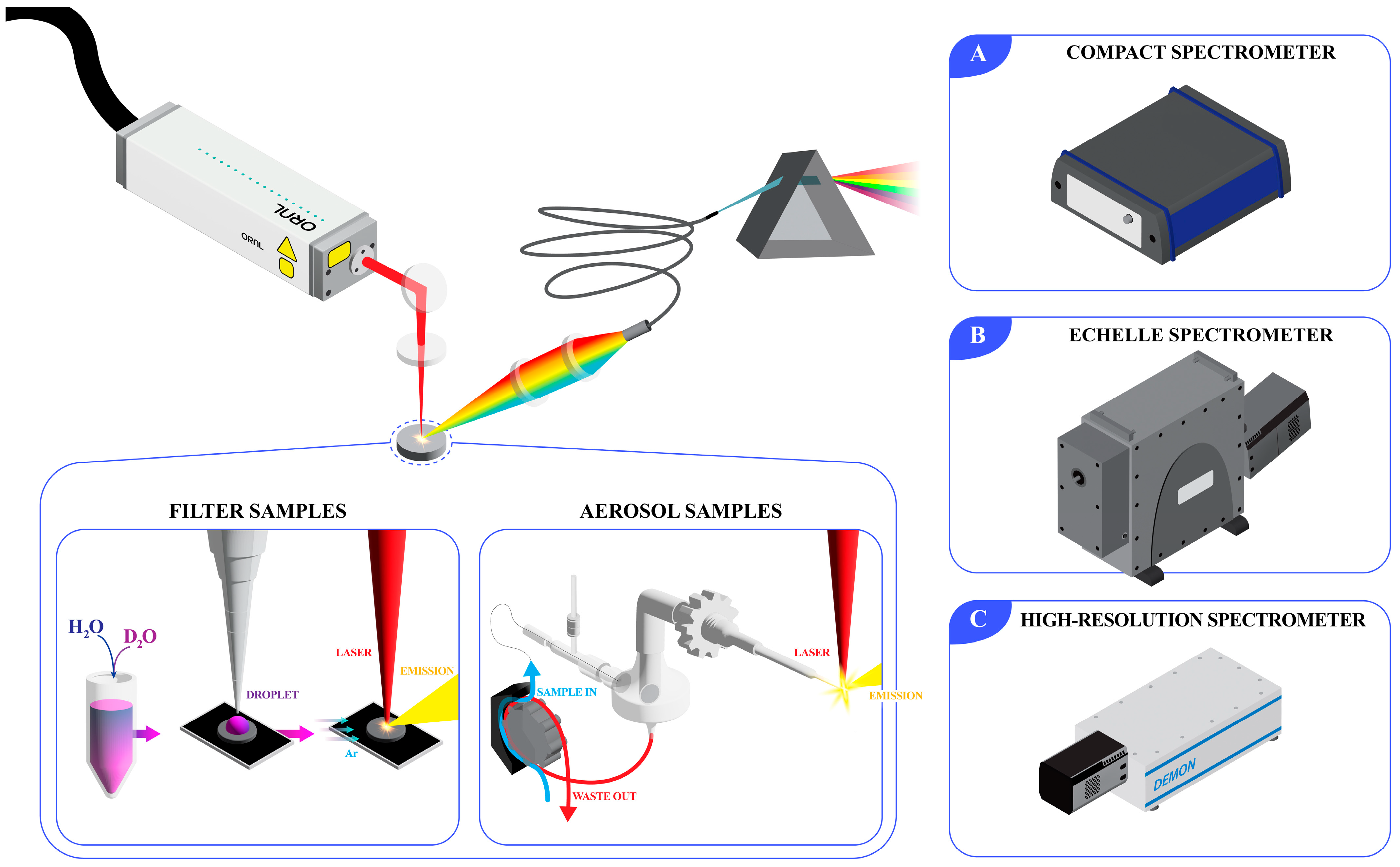
3.1. Comparison of Spectrometer Types
The first goal of this study was to demonstrate the effect that spectrometer resolution and type have on the measurement of hydrogen isotopes. When developing LIBS systems for online monitoring applications, it is important to consider the balance between cost, resolution, sensitivity, and spectral coverage when selecting a spectrometer. Here, three spectrometers have been compared: (1) a compact spectrometer that is typically used in a set of six or eight to provide full coverage of the 190–1000 nm wavelength range at a low resolution; (2) an echelle-type spectrometer equipped with an ICCD, which provides better sensitivity and moderate resolution with full wavelength coverage; and (3) a double echelle monochromator spectrometer, which provides top-tier resolution but only covers a small wavelength window. The costs associated with each spectrometer increases from option 1 to 3. Each spectrometer was used to measure the same set of calibration samples and their spectra, and their corresponding predictive capabilities were also compared.
Performing LIBS directly on liquids can be difficult because of plasma shockwave propagation effects [
18]. The shockwave that is generated with each laser pulse causes liquids to splash, risking the integrity of optical components. The subsequent wake leaves the liquid surface a variable distance from the optimal laser focal point. Although these issues can be counteracted through engineered sampling approaches, that is not the purpose of this study. Based on this knowledge, the initial hydrogen isotope measurements were performed by saturating glass fiber filters with aliquots of varying isotopic ratios and rastering the filter. Based on the scan pattern selected, there were 64 shots per sample. The hydrogen 656 nm emission line experiences significant Stark broadening, making its peak width extend over several nanometers. Here, Stark broadening refers to the broadening of emission lines due to the interaction between the excited species and the plasma itself. For many species, this broadening mechanism can be reduced by observing later periods in the plasma lifetime, but the hydrogen emission is short-lived, and the broadening cannot be avoided at atmospheric pressures. Single-shot emissions are typically very broad and subject to noise [
9]. To overcome this issue, several shots were averaged so that the peak center could be better resolved.
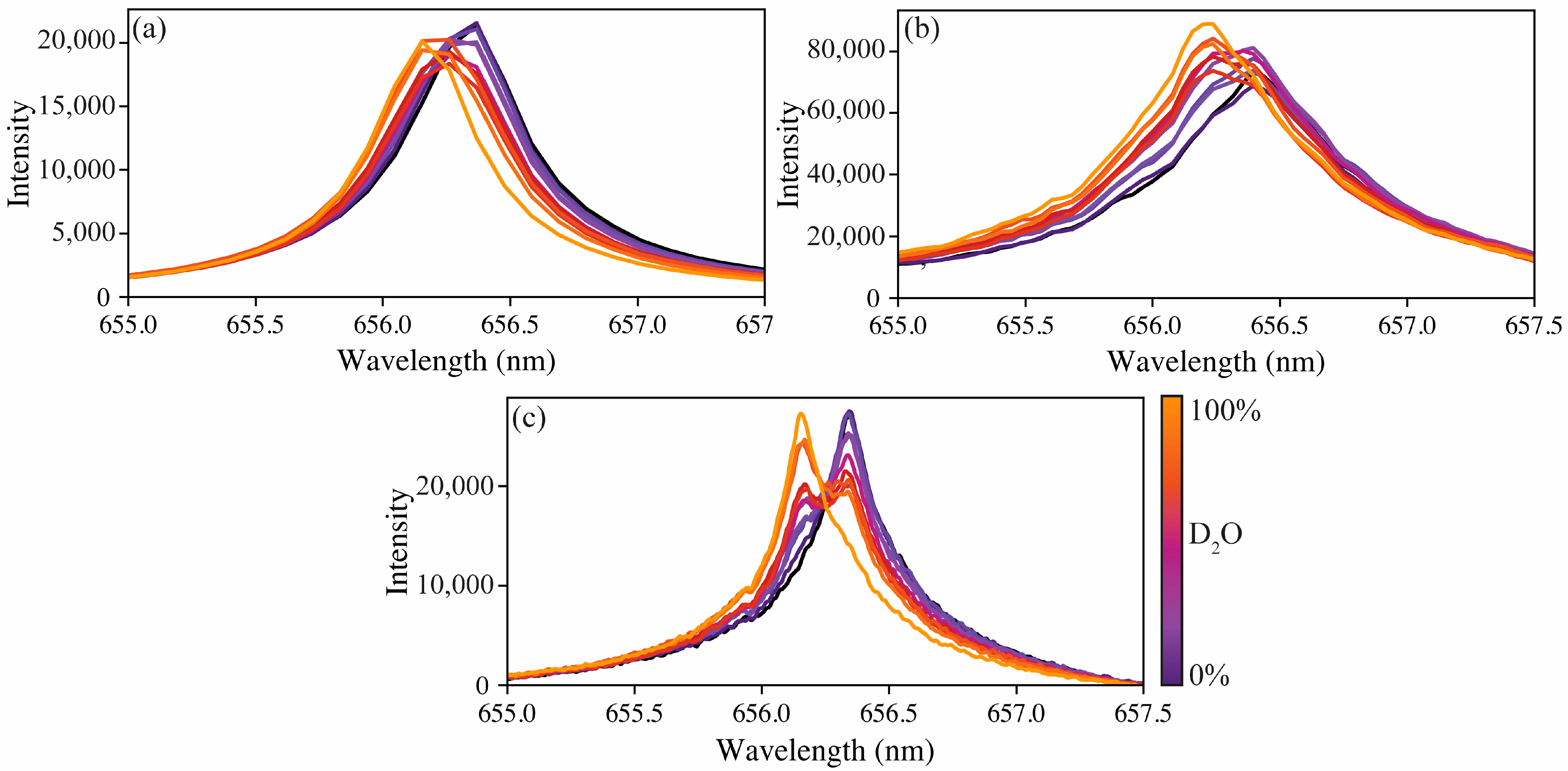
Figure S1 demonstrates the variation found in single-shot spectra versus the averaged spectra. Although fewer shots can be averaged, it was found that averaging all 64 shots provided the best spectra for modeling. The averaged spectra for the calibration sets for each spectrometer are shown in
Figure 2.
The difference in spectral resolution is apparent when looking at
Figure 2. Although all three spectrometer types can measure the peak shift, the resolution of the compact spectrometer means the protium and deuterium emissions are only separated by one wavelength step. The echelle spectrometer better captures the change from protium to deuterium; however, as anticipated, the high-resolution spectrometer captures these changes the best. The apparent peak shifts were measured to be 215, 158, and 186 pm to blue for the compact, echelle, and high-resolution spectrometers, respectively. The literature-reported peak shift from protium to deuterium is approximately 180 pm [
9]. For tritium measurements, the emission peak would be shifted further towards the blue due to the increased mass difference, [
19] The blue shift between tritium and deuterium is smaller than deuterium and protium (60 pm vs. 180 pm, respectively) [
19]. Fortunately in an MSR, tritium and protium are would be the most abundant hydrogen isotopes so issues related to deconvoluting deuterium and tritium emissions will be limited [
1].
Because the isotope shift can be seen on each of the spectrometers, the pertinent question is the ability of the spectrometers to quantify the protium-to-deuterium ratio difference. With this goal in mind, several chemometric methods were used to construct models to quantify the isotope ratio. Here, models were built using PCR, PLSR, and MCR [
20,
21,
22,
23,
24].
PCR is performed by first applying principal component analysis (PCA) to reduce the dimensionality of the spectral dataset. PCA identifies orthogonal vectors that explain the variance of the independent variable (spectra) and then reduces the dataset to scores for each principal component. PCR then applies ordinary linear regression to these PCA scores [
23]. PLSR considers the signal matrix (spectra) and the response matrix (isotope ratio) and transforms them into a latent space [
21,
22]. Here, latent variables that explain the most covariance between the signal and response matrices are solved iteratively. This process is similar to PCA; however, PCA seeks to explain the most variance in the signal matrix, whereas PLSR seeks to explain the most covariance between the signal and response matrices. In this study, only two principal components or latent variables were used for PCR and PLSR models, respectively. The third method used was MCR, which refers to the use of an alternating least squares approach to resolving the pure components from a mixture [
24]. Here, MCR was applied with a non-negative constraint and a normality constraint dictating that all component concentrations sum to unity. MCR was provided with the calibration dataset and the corresponding response matrix, with which MCR determined the two spectral signals corresponding to pure protium or deuterium. MCR can be used for prediction by fitting the pure spectra components to measured spectra. Here, PCR represents the simplest modeling approach. PLSR typically provides superior prediction capabilities compared to PCR and is the most common multivariate model used for LIBS spectral analysis. Lastly, MCR represents an alternative model which provides superior interpretability by providing the pure component spectra.
To evaluate model performance, cross-validation was performed using a leave-one-out cross-validation (LOOCV) approach. Here, the model was iteratively built, leaving one sample out at a time, and then, at each iteration, the sample left out was used to test the model. The residuals for each sample while they are left out are used to calculate the root mean square error of cross-validation (RMSECV):
where
yi is the known concentration value of the
ith sample left out during the LOOCV iteration,
is the model-predicted concentration, and
n is the number of samples [
25].
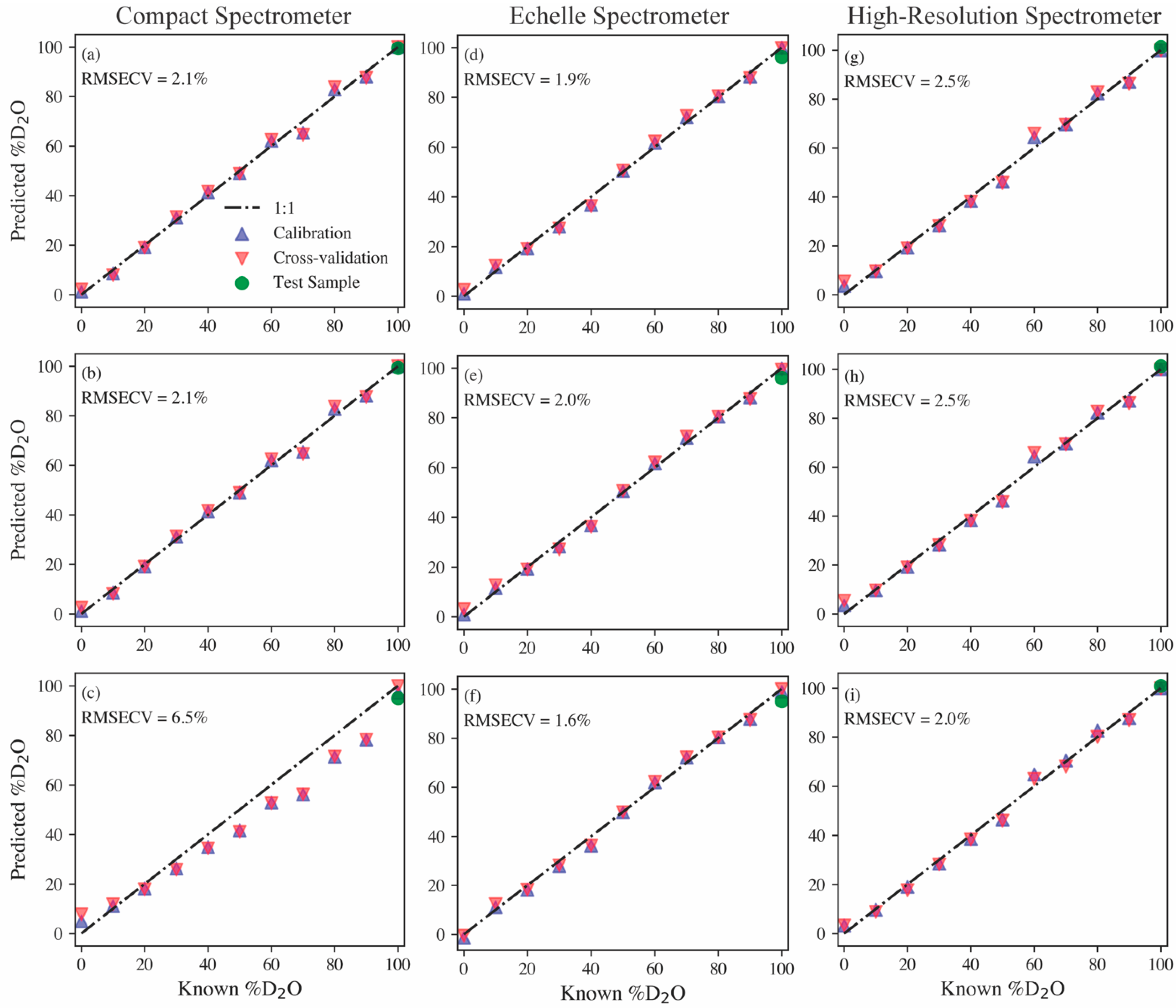
Parity plots for PCR, PLSR, and MCR models built using each spectrometer type, along with their RMSECV values, are shown in
Figure 3. The optimal models were built after minor preprocessing. Firstly, the high-resolution and echelle spectra were smoothed using a Savitzky–Golay filter with a first-order polynomial and a five-point window. Next, each spectrum was baseline-adjusted by subtracting the average background levels near the hydrogen peak. Lastly, each spectrum was normalized to the maximum intensity between 650 and 665 nm. The PCR and PLSR models provide relatively similar prediction performances regardless of the spectrometer type, with RMSECVs ranging from 1.9% to 2.5%. The MCR model results provide insight into how well the protium and deuterium emissions can be deconvoluted from one another based on the spectrometer. Examples of PCA loadings, PLSR regression coefficients, and the MCR pure spectral components from the echelle models are shown in the
Supporting Information (Figures S2–S4). The compact spectrometer clearly struggles here with an RMSECV of 6.5%. The echelle and high-resolution spectrometer MCR models have RMSECV values on par with the previously discussed PCR and PLSR models, but the echelle MCR model performs the best out of all multivariate models with an RMSECV of 1.6%. Although
Figure 2 shows the high-resolution spectrometer, providing a clearer distinguishable isotope shift, these model results indicate that the echelle spectrometer with a lower resolution can provide equivalent, if not better, quantification of the hydrogen isotope ratio.
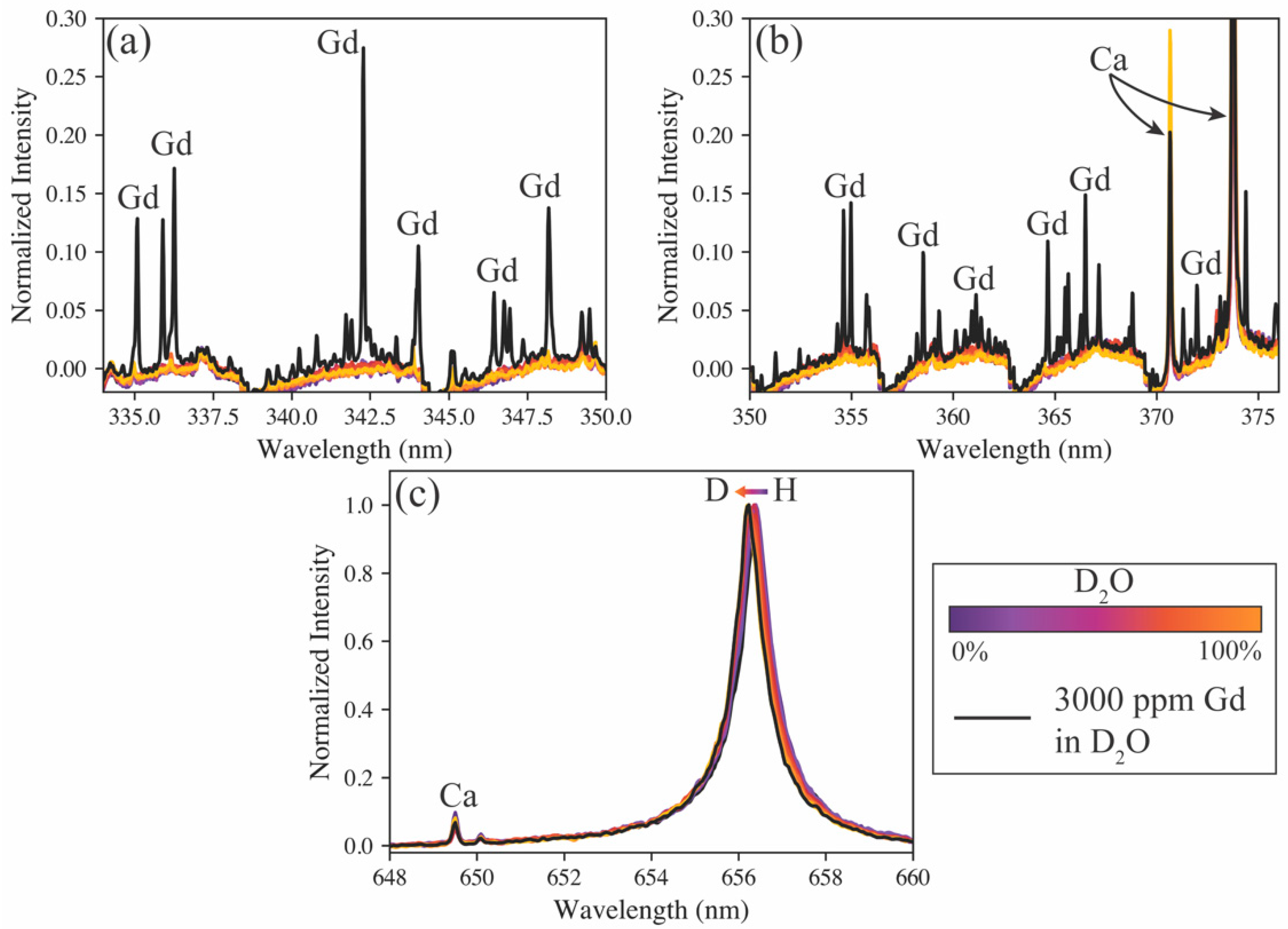
A sample containing 3000 ppm gadolinium in D
2O was used as a test of the models’ predictive capabilities on samples doped with other species. As seen in
Figure 3 (green markers), every model estimates the hydrogen isotope ratio to be nearly 100% deuterium, as expected. This sample also provides insight into the versatility of the various spectrometers. For example, the high-resolution spectrometer may accurately monitor the hydrogen isotope ratio, but it is blind to the additional species added to the sample. Conversely, the echelle spectrometer can measure the hydrogen isotope ratio, as well as measure the added gadolinium emissions (see
Figure 4). Gadolinium as the test sample addition demonstrates this capability well because of its large number of emission peaks, representative of many lanthanides, which are fission products and, in some cases, neutron poisons in MSRs [
4]. The compact spectrometer has the same benefit of measuring more than just the hydrogen emissions, but in this case, the gadolinium emissions fell beyond the window of the spectrometer used. Typically, when compact spectrometers are used, several are used simultaneously; each spectrometer monitors different wavelength ranges, and their spectra can be stitched together to provide a broadband spectrum. Based on model performance and the ability to measure isotope shifts and additional species simultaneously, the echelle spectrometer was selected for further tests on aerosol samples.
3.2. Transferring Filter Calibration to Aerosol Measurements
The off-gas system of an MSR is a major pathway for fission product and decay daughter removal from the core. An aerosol stream is anticipated to be formed regardless of if the fuel salt is sparged or simply swept with a cover gas. Based on this expectation, it was important to demonstrate the ability to monitor hydrogen isotopes via LIBS in a continuous aerosol stream. In addition to this demonstration, the experiment poses a challenge that would be realistic for a deployed LIBS system: how a model can be effectively calibrated without running a full set of calibration samples in situ.
Firstly, the same 11 mixtures of H
2O and D
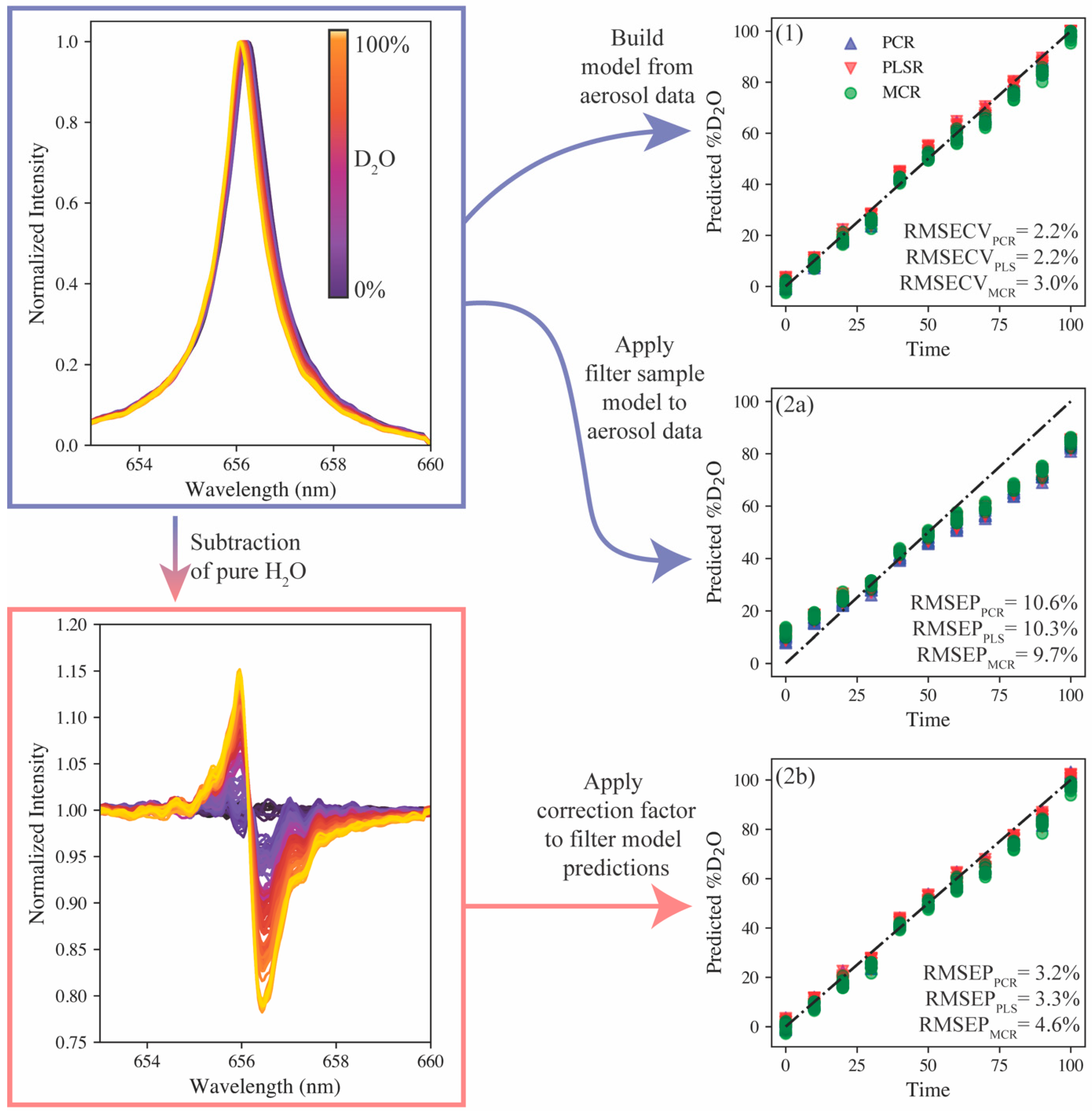
2O were run through the aerosol introduction system and 10 accumulate spectra (100 shots each) were collected. The normalized hydrogen emission and corresponding peak shift is shown in
Figure 5. The hydrogen emission visually resembles that shown in
Figure 4c; however, the broadening of the emission is larger than that seen on the filter sample set. This difference is the result of the change in matrix; the plasma formed in the aerosol stream had a greater electron density than the filter samples. The PCR, PLSR, and MCR models, with the same preprocessing steps discussed previously, were reconstructed using the aerosol spectra. The results are shown in
Figure 5 (model 1). The RMSECV values were calculated to be 2.2%, 2.2%, and 3.0% for the PCR, PLSR, and MCR models, respectively. These values are larger than those using the filter samples analyzed by the echelle spectrometer, but this difference can likely be attributed to increased peak broadening that reduces the resolution of the protium and deuterium emissions.
Next, owing to the similar modeling approaches for the filter and aerosol sample sets, an attempt was made to use the chemometric models calibrated on the filter samples to predict the aerosol hydrogen isotopic compositions. The parity plot for these models’ predictions is shown in
Figure 5 (model 2a). Here, because the parity plots show data not used in the calibration, the prediction error metric is the root mean square error of prediction (RMSEP). The RMSEP values range between 9.7% and 10.6%. The parity plot shows the predictions falling into tight groupings and the overall prediction series still being linear, albeit at a slope of less than 1. This is a direct consequence of the difference in sample matrix (i.e., aerosol vs. filters). While many matrix effects are reduced using preprocessing steps, the differences in Stark broadening due to differences in the plasma electron density still impact the application of the filter model onto the aerosol matrix.
To adjust the filter sample models to better predict the aerosol data, the pure protium emission spectra were subtracted from the entire filter training set, and the model was rebuilt. The aerosol data were modified in the same way before applying the model for predictions (see
Figure 5). This modification forced the two datasets (calibration and prediction) to collapse onto one another, but another correction was needed to correct the slope deviation on the parity plot. For this correction, the models’ prediction of the pure deuterium sample was used to generate a correction factor to apply to all model predictions. The factor was calculated to be 1.35. The predictions of the adjusted models with the correction factor applied are shown in
Figure 5 (model 2b). The RMSEP values were calculated to be 3.2%, 3.3%, and 4.6% for PCR, PLSR, and MCR, respectively. These values are greater than the model trained directly on the aerosol samples, but the PCR and PLSR models would still be valuable for quantitative monitoring. The approach of using the pure protium and deuterium aerosol samples to adjust the filter sample set model for aerosol predictions represents an 82% reduction in the volume of sample used compared with a model trained entirely on aerosol samples. This approach also represents a more feasible approach to training isotopic LIBS models for deployed systems by only needing the model end points (each pure isotope) to be tested in situ versus running a series of samples in the field.
3.3. Real-Time Monitoring Demonstration
As a final test and demonstration of the utility of the constructed models, a real-time test was conducted. This test leveraged the aerosol sample introduction system to pump solutions from a reservoir where the sample stream could be modified in real time by spiking, diluting, or completely changing the reservoir liquid while measuring LIBS spectra continuously. In an effort to show greater applicability to MSR effluent streams, FLiNaK salt (47:11:42 mol% LiF:NaF:KF) was dissolved in either H2O or D2O to serve as spikes during the test. The ability of LIBS to monitor each of these salt species along with the hydrogen isotope ratio truly highlights the technique’s versatility for MSR applications.
The real-time test was performed as follows. Firstly, pure H
2O was run to establish a baseline, then at
t1 (~3.8 min), the first spike of FLiNaK in D
2O was added to the reservoir. This mixture was run until
t2 (~7.5 min), when a spike of FLiNaK in H
2O was added to the reservoir. At
t3 (~10.2 min), a spike of pure H
2O was added to dilute both salt species and the hydrogen isotope ratio. Lastly, at
t4 (~13.6 min), the entire reservoir was replaced with pure H
2O to return to the baseline. LIBS spectra were recorded in 100 shot accumulates at 10 Hz, providing 100 spectra over the 16.6 min. The results of the real-time test are shown in
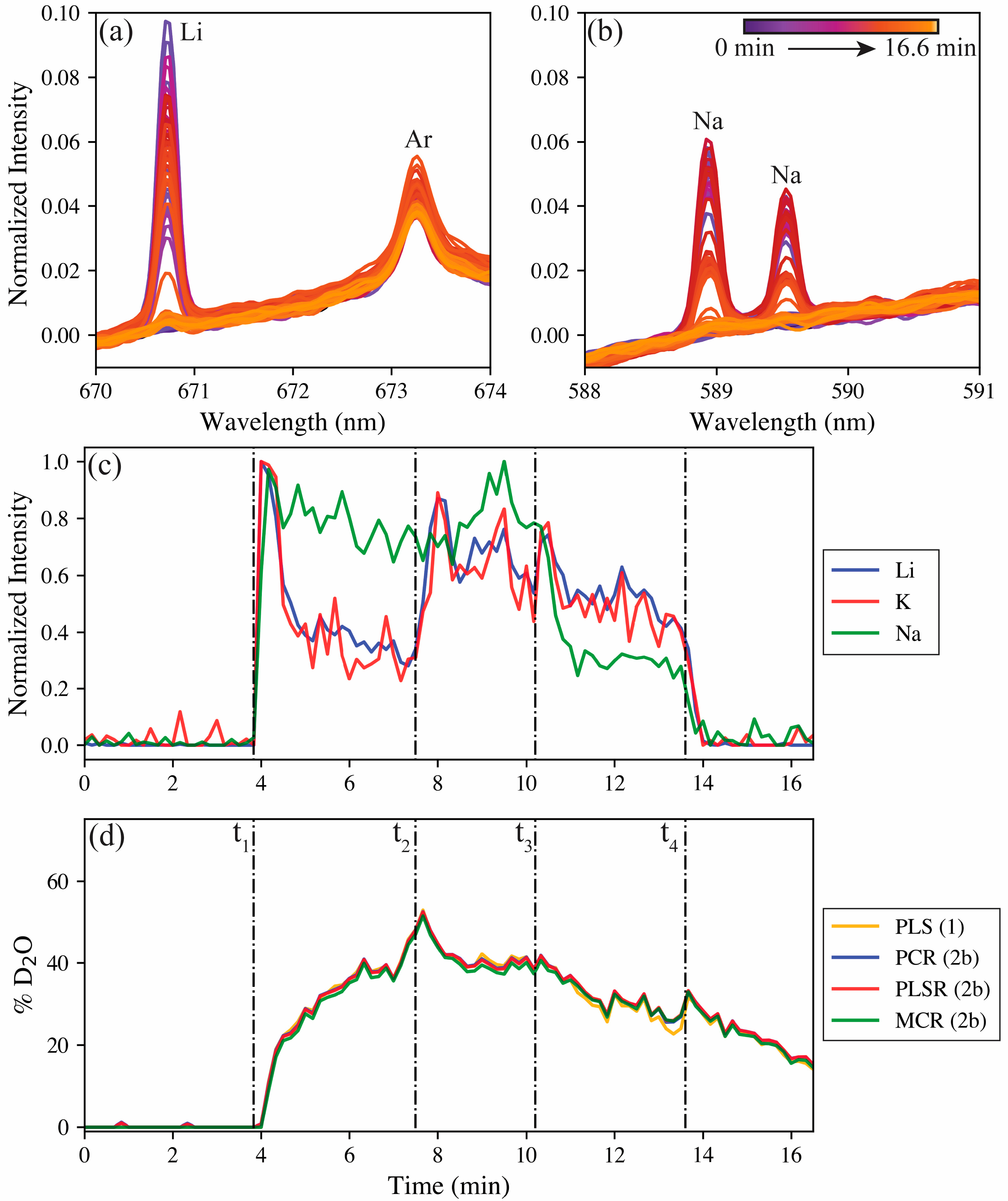
Figure 6.
The spectral response of the Li 670.8 nm emission and the Na 589 and 589.59 nm doublet are shown in
Figure 6a,b. Both are nonexistent at the beginning (purple), but they rise and fall during the test from the changing reservoir before returning to the baseline at the end (bright yellow) of the test. The Ar I 763.2 nm emission remains relatively constant during the test because it originates from the aerosol carrier gas.
Figure 6c shows the normalized trends for lithium (670.8 nm), sodium (589 nm), and potassium (693.9 nm, see
Figure S5) over the duration of the tests, with the spike time stamps indicated by the dashed lines. Similarly, the model predictions for the hydrogen isotope ratio based on changes in peak position in the real-time test are shown in
Figure 6d. No meaningful signals are seen for any of the salt species before their levels rapidly elevate following the first spike at
t1. Following the initial spike, the lithium and potassium levels begin to decay. The decay of lithium and potassium can be attributed to the spike at
t1 being a supersaturated mixture, where the LiF and KF crash out of solution after the initial mixture has time to settle in the reservoir. The LiF makes up 30 wt% of the FLiNaK salt composition and has a low solubility in water, so this behavior is not unexpected. The KF accounts for 59 wt% of the FLiNaK composition, so despite its high solubility in water, the amount present in the spike was above the solubility limit. The NaF accounts for far less of the FLiNaK composition (11 wt%); with this lesser amount, combined with its solubility being just under half that of KF, the NaF was expected to be nearly fully dissolved in solution, which explains its differing behavior from the other salt species. Following the D
2O spike at
t1, the hydrogen isotope ratio slowly shifts from 0% to approximately 50% deuterium, as expected, with the spike doubling the reservoir volume. The slow ingrowth of deuterium indicates that latency does exist in the aerosol sampling system with regards to the hydrogen isotopes.
The second spike contains the same concentration of FLiNaK in H2O. Following this spike at t2, the salt levels again spike before decaying away. Here, the hydrogen isotope ratio begins to shift back toward protium, and following the spike of pure H2O at t3, the hydrogen isotope ratio continues to decay at a similar rate. After this third spike, the sodium levels drop owing to being diluted by the additional H2O; however, the lithium and potassium levels spike and then return to a similar level as before the third spike. This spike is because of lithium and potassium salts not in solution in the reservoir now being able to dissolve to maintain the solubility limit in the reservoir. Lastly, the reservoir is completely changed to pure H2O at t4, the salt levels plummet to zero, and the hydrogen isotope ratio continues its decay back toward protium.
The real-time test offered the valuable opportunity to compare the multivariate models trained on the aerosol calibration samples and the filter sample models after they were corrected for aerosol measurements. The corrected PCR, PLSR, and MCR model predictions are shown in
Figure 6d, overlaid with the predictions of the all-aerosol trained PLSR model. Interestingly, despite the RMSE differences between the models (see
Figure 5), the disparities between model performance during the real-time test was miniscule. This difference further boasts of the opportunity offered by the ability to train LIBS models on one medium and then correct for systematic differences to transfer that model to the in-situ testing setup.
Overall, the real-time tests highlight the versatility of LIBS as an elemental/isotopic online monitoring tool for MSRs and other industrial applications. Here, LIBS monitored three independent elements that are directly tied to MSRs as typical salt species. Despite the intent to vary these salt species in similar profiles, LIBS was able to detect impacts of solubility on the true salt species composition in the aerosol stream. Meanwhile the transferred filter calibration models were able to track hydrogen isotopes with little difference compared to the all-aerosol trained system. This reaffirms that isotopic LIBS models can be built on benchtop systems and then transferred to alternative systems (e.g., engineering scale tests) by testing only two samples at the sampling destination. This will be particularly useful when these in situ calibration tests are performed under time-restrictions, such as in radioactive environments, and when the isotopes needed to train the models are limited. Being able to simultaneously track element profiles and specific isotopes (e.g., 3H, 235U, 238U) will be vital for MSR monitoring.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}