
Polarization Conforms Performance Variability in Amorphous Electrodeposited Iridium Oxide pH Sensors: A Thorough Surface Chemistry Investigation

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Substrate Fabrication
2.2. Electrodeposition Process
2.3. Methods
2.4. XPS Experimental Flow
3. Results and Discussion
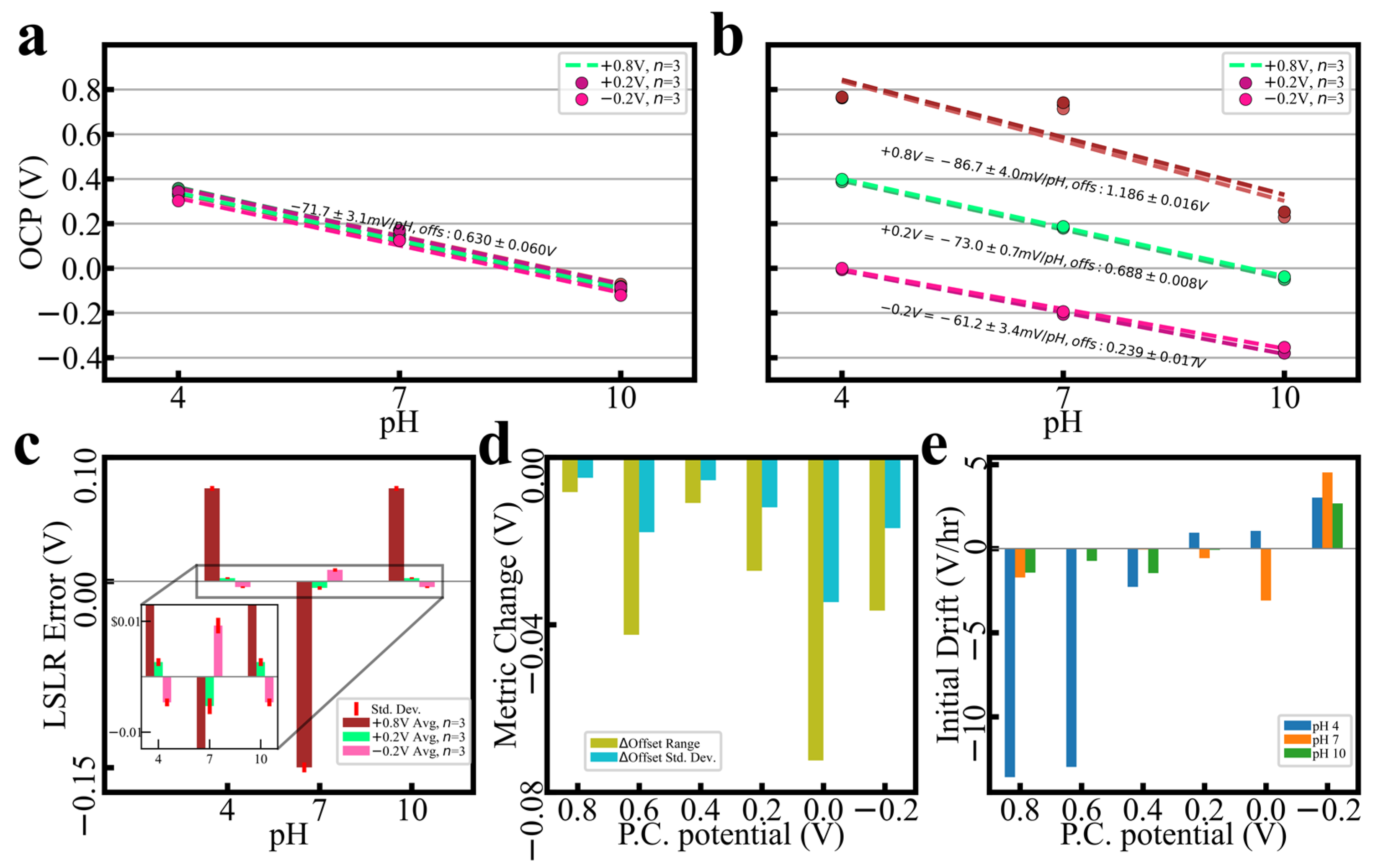
3.1. PC Performance Control
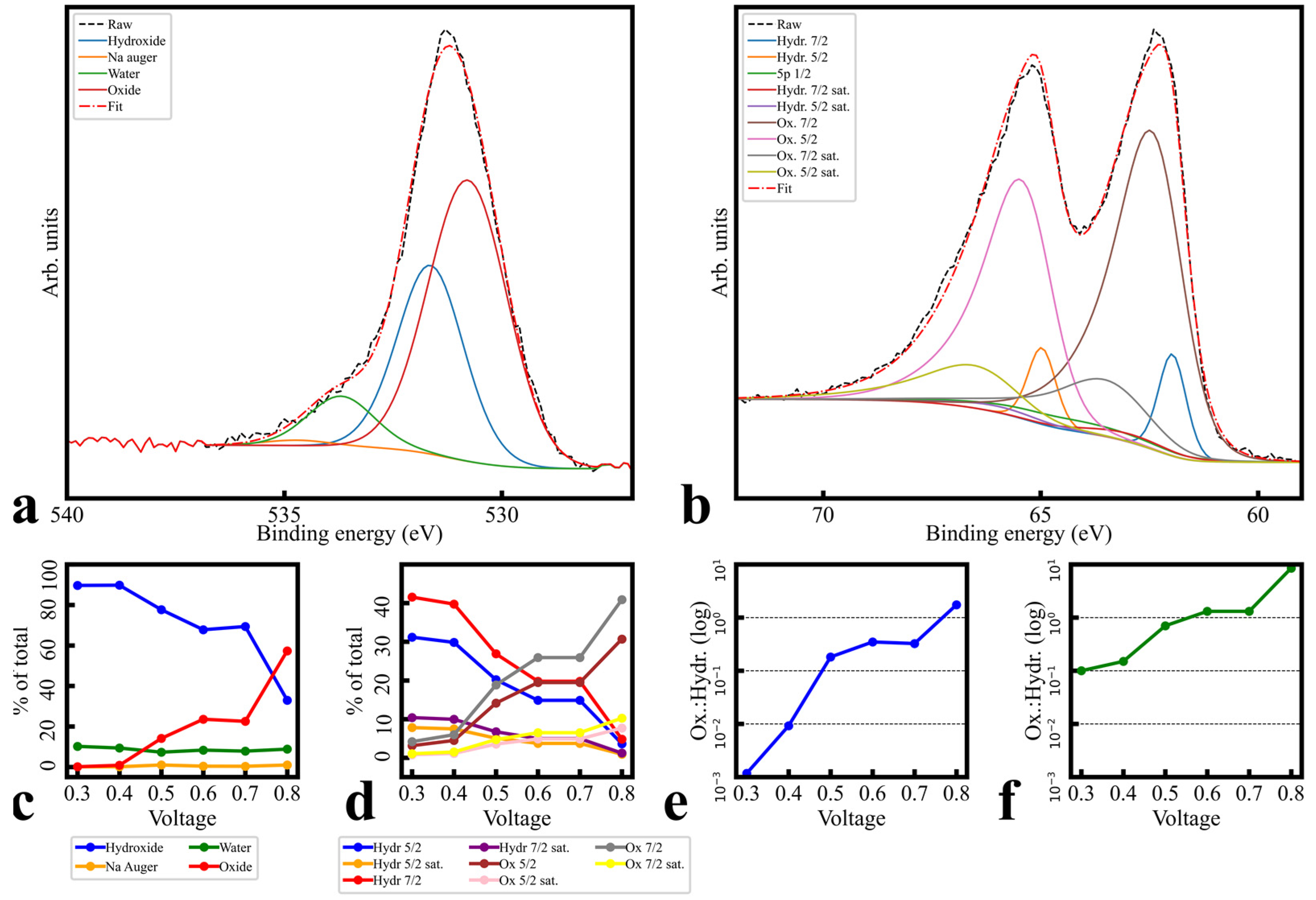
3.2. Morphology and Elemental Composition
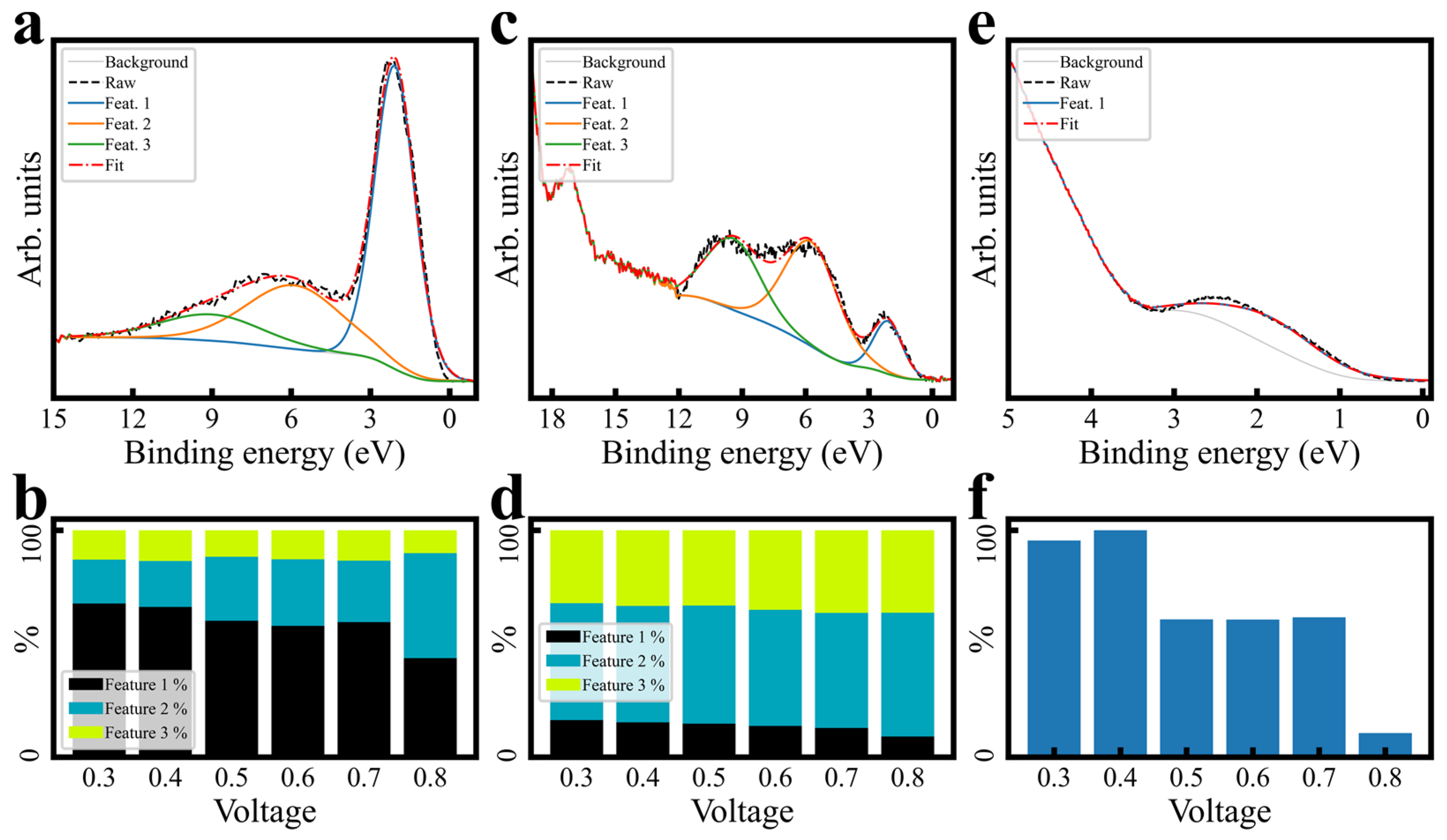
3.3. Surface Bonding Investigation
3.3.1. Raw Polarization Trends
3.3.2. Fitting and Quantification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghoneim, M.; Nguyen, A.; Dereje, N.; Huang, J.; Moore, G.; Murzynowski, P.; Dagdeviren, C. Recent Progress in Electrochemical pH-Sensing Materials and Configurations for Biomedical Applications. Chem. Rev. 2019, 119, 5248–5297. [Google Scholar] [CrossRef]
- Olthuis, W.; Robben, M.; Bergveld, P.; Bos, M.; Van Der Linden, W. pH sensor properties of electrochemically grown iridium oxide. Sens. Actuators B Chem. 1990, 2, 247–256. [Google Scholar] [CrossRef]
- Amor, H.E.; Kouki, A.B.; Marsh, P.; Kim, K.T.; Cao, H. Development of a novel miniaturized LTCC-based wireless pH sensing system. In Proceedings of the 2016 IEEE SENSORS, Orlando, FL, USA, 30 October–3 November 2016; pp. 1–3. [Google Scholar]
- Marsh, P.; Manjakkal, L.; Yang, X.; Huerta, M.; Le, T.; Thiel, L.; Chiao, J.-C.; Cao, H.; Dahiya, R. Flexible Iridium Oxide Based pH Sensor Integrated With Inductively Coupled Wireless Transmission System for Wearable Applications. IEEE Sens. J. 2020, 20, 5130–5138. [Google Scholar] [CrossRef]
- Marsh, P.; Zhuang, Y.; Xu, Z.; Heine, L.; Cao, H. Sample Tube pH Monitoring via Passive Powering and Communication. In Proceedings of the 2019 IEEE SENSORS, Montreal, QC, Canada, 27–30 October 2019; pp. 1–4. [Google Scholar]
- Carroll, S.; Baldwin, R.P. Self-calibrating microfabricated iridium oxide pH electrode array for remote monitoring. Anal. Chem. 2010, 82, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Elsen, H.A. Thermodynamic and Dynamic Investigations of Hydrated Iridium Oxide Potentiometric PH Micro-Sensors; University of California: Berkeley, CA, USA, 2007. [Google Scholar]
- Cao, H.; Landge, V.; Tata, U.; Seo, Y.-S.; Rao, S.; Tang, S.-J.; Tibbals, H.; Spechler, S.; Chiao, J.-C. An implantable, batteryless, and wireless capsule with integrated impedance and pH sensors for gastroesophageal reflux monitoring. IEEE Trans. Biomed. Eng. 2012, 59, 3131–3139. [Google Scholar] [PubMed]
- Cao, H.; Nguyen, C.; Chiao, J. Fabrication and surface-modification of implantable microprobes for neuroscience studies. Adv. Nat. Sci. Nanosci. Nanotechnol. 2012, 3, 025003. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Velasco Vélez, J.J.; Massué, C.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Greiner, M.T.; Allan, J. The electronic structure of iridium and its oxides. Surf. Interface Anal. 2016, 48, 261–273. [Google Scholar] [CrossRef]
- Freakley, S.J.; Ruiz-Esquius, J.; Morgan, D.J. The X-ray photoelectron spectra of Ir, IrO2 and IrCl3 revisited. Surf. Interface Anal. 2017, 49, 794–799. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Li, D.; Batchelor-McAuley, C.; Compton, R.G. A simplified methodology: pH sensing using an in situ fabricated Ir electrode under neutral conditions. J. Solid State Electrochem. 2021, 25, 2821–2833. [Google Scholar] [CrossRef]
- Tolosa, V.M.; Wassum, K.M.; Maidment, N.T.; Monbouquette, H.G. Electrochemically deposited iridium oxide reference electrode integrated with an electroenzymatic glutamate sensor on a multi-electrode arraymicroprobe. Biosens. Bioelectron. 2013, 42, 256–260. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, X.; Ye, Z.; Zhang, J.; Cao, F.; Zhang, J. A fabrication of iridium oxide film pH micro-sensor on Pt ultramicroelectrode and its application on in-situ pH distribution of 316L stainless steel corrosion at open circuit potential. Sens. Actuators B Chem. 2018, 255, 1974–1982. [Google Scholar] [CrossRef]
- VanHoudt, P.; Lewandowski, Z.; Little, B. Iridium oxide pH microelectrode. Biotechnol. Bioeng. 1992, 40, 601–608. [Google Scholar] [CrossRef]
- Hitchman, M.L.; Ramanathan, S. Evaluation of iridium oxide electrodes formed by potential cycling as pH probes. Analyst 1988, 113, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.; Nielsen, R.J.; Goddard III, W.A. The reaction mechanism with free energy barriers at constant potentials for the oxygen evolution reaction at the IrO2 (110) surface. J. Am. Chem. Soc. 2017, 139, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hendrikse, J.; Olthuis, W.; Bergveld, P. A method of reducing oxygen induced drift in iridium oxide pH sensors. Sens. Actuators B Chem. 1998, 53, 97–103. [Google Scholar] [CrossRef]
- Cherevko, S.; Geiger, S.; Kasian, O.; Mingers, A.; Mayrhofer, K.J. Oxygen evolution activity and stability of iridium in acidic media. Part 2.–Electrochemically grown hydrous iridium oxide. J. Electroanal. Chem. 2016, 774, 102–110. [Google Scholar] [CrossRef]
- Scohy, M.; Abbou, S.; Martin, V.; Gilles, B.; Sibert, E.; Dubau, L.; Maillard, F. Probing Surface Oxide Formation and Dissolution on/of Ir Single Crystals via X-ray Photoelectron Spectroscopy and Inductively Coupled Plasma Mass Spectrometry. ACS Catal. 2019, 9, 9859–9869. [Google Scholar] [CrossRef]
- Zagalskaya, A.; Alexandrov, V. Mechanistic Study of IrO2 Dissolution during the Electrocatalytic Oxygen Evolution Reaction. J. Phys. Chem. Lett. 2020, 11, 2695–2700. [Google Scholar] [CrossRef]
- Steegstra, P.; Ahlberg, E. Influence of oxidation state on the pH dependence of hydrous iridium oxide films. Electrochim. Acta 2012, 76, 26–33. [Google Scholar] [CrossRef]
- Steegstra, P.; Busch, M.; Panas, I.; Ahlberg, E. Revisiting the redox properties of hydrous iridium oxide films in the context of oxygen evolution. J. Phys. Chem. C 2013, 117, 20975–20981. [Google Scholar] [CrossRef]
- Marsh, P.; Mohseni, F.; Chiao, J.-C.; Cao, H. Investigation of the Self-Calibration Function for IrOx-based pH Sensors. In Proceedings of the 2020 IEEE SENSORS, Rotterdam, The Netherlands, 25–28 October 2020; pp. 1–4. [Google Scholar]
- Elsen, H.A.; Monson, C.F.; Majda, M. Effects of electrodeposition conditions and protocol on the properties of iridium oxide pH sensor electrodes. J. Electrochem. Soc. 2009, 156, F1–F6. [Google Scholar] [CrossRef]
- Yamanaka, K. Anodically electrodeposited iridium oxide films (AEIROF) from alkaline solutions for electrochromic display devices. Jpn. J. Appl. Phys. 1989, 28, 632. [Google Scholar] [CrossRef]
- Burke, L.; Scannell, R. The effect of UV light on the hydrous oxides of iridium. J. Electroanal. Chem. Interfacial Electrochem. 1988, 257, 101–121. [Google Scholar] [CrossRef]
- Smith, R.D.; Sporinova, B.; Fagan, R.D.; Trudel, S.; Berlinguette, C.P. Facile photochemical preparation of amorphous iridium oxide films for water oxidation catalysis. Chem. Mater. 2014, 26, 1654–1659. [Google Scholar] [CrossRef]
- Kim, D.-H.; Park, S.-H.; Choi, J.; Yi, M.H.; Kim, H.-S. Fabrication of iridium oxide nanoparticles supported on activated carbon powder by flashlight irradiation for oxygen evolutions. Mater. Sci. Eng. B 2015, 201, 29–34. [Google Scholar] [CrossRef]
- Hämäläinen, J.; Kemell, M.; Munnik, F.; Kreissig, U.; Ritala, M.; Leskelä, M. Atomic layer deposition of iridium oxide thin films from Ir (acac) 3 and ozone. Chem. Mater. 2008, 20, 2903–2907. [Google Scholar] [CrossRef]
- Kötz, R.; Neff, H.; Stucki, S. Anodic Iridium Oxide Films: XPS-Studies of Oxidation State Changes and. J. Electrochem. Soc. 1984, 131, 72. [Google Scholar] [CrossRef]
- Gaarenstroom, S.; Winograd, N. Initial and final state effects in the ESCA spectra of cadmium and silver oxides. J. Chem. Phys. 1977, 67, 3500–3506. [Google Scholar] [CrossRef]
- Kötz, E.; Neff, H. Anodic iridium oxide films: An UPS study of emersed electrodes. Surf. Sci. 1985, 160, 517–530. [Google Scholar] [CrossRef]
- Riga, J.; Tenret-Noel, C.; Pireaux, J.-J.; Caudano, R.; Verbist, J.; Gobillon, Y. Electronic structure of rutile oxides TiO2, RuO2 and IrO2 studied by X-ray photoelectron spectroscopy. Phys. Scr. 1977, 16, 351. [Google Scholar] [CrossRef]
- Ping, Y.; Galli, G.; Goddard III, W.A. Electronic structure of IrO2: The role of the metal d orbitals. J. Phys. Chem. C 2015, 119, 11570–11577. [Google Scholar] [CrossRef]
- NIST X-ray Photoelectron Spectroscopy Database. NIST Standard Reference Database Number 20. 2000. Available online: https://srdata.nist.gov/xps/ (accessed on 21 December 2023).
- Lervik, I.A.; Tsypkin, M.; Owe, L.-E.; Sunde, S. Electronic structure vs. electrocatalytic activity of iridium oxide. J. Electroanal. Chem. 2010, 645, 135–142. [Google Scholar] [CrossRef]
- Kawar, R.; Chigare, P.; Patil, P. Substrate temperature dependent structural, optical and electrical properties of spray deposited iridium oxide thin films. Appl. Surf. Sci. 2003, 206, 90–101. [Google Scholar] [CrossRef]
- Greczynski, G.; Hultman, L. Reliable determination of chemical state in X-ray photoelectron spectroscopy based on sample-work-function referencing to adventitious carbon: Resolving the myth of apparent constant binding energy of the C 1s peak. Appl. Surf. Sci. 2018, 451, 99–103. [Google Scholar] [CrossRef]
- Dupin, J.-C.; Gonbeau, D.; Vinatier, P.; Levasseur, A. Systematic XPS studies of metal oxides, hydroxides and peroxides. Phys. Chem. Chem. Phys. 2000, 2, 1319–1324. [Google Scholar] [CrossRef]
- McCafferty, E.; Wightman, J. Determination of the concentration of surface hydroxyl groups on metal oxide films by a quantitative XPS method. Surf. Interface Anal. 1998, 26, 549–564. [Google Scholar] [CrossRef]
- Cruz, A.; Abad, L.; Carretero, N.; Moral-Vico, J.; Fraxedas, J.; Lozano, P.; Subías, G.; Padial, V.; Carballo, M.; Collazos-Castro, J. Iridium oxohydroxide, a significant member in the family of iridium oxides. Stoichiometry, characterization, and implications in bioelectrodes. J. Phys. Chem. C 2012, 116, 5155–5168. [Google Scholar] [CrossRef]
- Yano, T.; Ebizuka, M.; Shibata, S.; Yamane, M. Anomalous chemical shifts of Cu 2p and Cu LMM Auger spectra of silicate glasses. J. Electron Spectrosc. Relat. Phenom. 2003, 131, 133–144. [Google Scholar] [CrossRef]
- Pavlovic, Z.; Ranjan, C.; Gao, Q.; van Gastel, M.; Schlogl, R. Probing the structure of a water-oxidizing anodic iridium oxide catalyst using Raman spectroscopy. ACS Catal. 2016, 6, 8098–8105. [Google Scholar] [CrossRef]
- Hong, D.; Murakami, M.; Yamada, Y.; Fukuzumi, S. Efficient water oxidation by cerium ammonium nitrate with [Ir III (Cp*)(4,4′-bishydroxy-2,2′-bipyridine)(H2O)] 2+ as a precatalyst. Energy Environ. Sci. 2012, 5, 5708–5716. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Vélez, J.J.V.; Arrigo, R.; Piccinin, S.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R. In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces. Chem. Sci. 2017, 8, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- Kahk, J.; Poll, C.; Oropeza, F.; Ablett, J.; Céolin, D.; Rueff, J.; Agrestini, S.; Utsumi, Y.; Tsuei, K.; Liao, Y. Understanding the Electronic Structure of IrO2 Using Hard-X-ray Photoelectron Spectroscopy and Density-Functional Theory. Phys. Rev. Lett. 2014, 112, 117601. [Google Scholar] [CrossRef] [PubMed]
- Bourlange, A.; Payne, D.; Palgrave, R.; Zhang, H.; Foord, J.; Egdell, R.; Jacobs, R.; Veal, T.; King, P.; McConville, C. The influence of Sn doping on the growth of In2O3 on Y-stabilized ZrO2 (100) by oxygen plasma assisted molecular beam epitaxy. J. Appl. Phys. 2009, 106, 013703. [Google Scholar] [CrossRef]
- Payne, D.J.; Egdell, R.G.; Hao, W.; Foord, J.S.; Walsh, A.; Watson, G.W. Why is lead dioxide metallic? Chem. Phys. Lett. 2005, 411, 181–185. [Google Scholar] [CrossRef]
- Sachse, R.; Pflüger, M.; Velasco-Vélez, J.-J.; Sahre, M.; Radnik, J.; Bernicke, M.; Bernsmeier, D.; Hodoroaba, V.-D.; Krumrey, M.; Strasser, P. Assessing Optical and Electrical Properties of Highly Active IrOx Catalysts for the Electrochemical Oxygen Evolution Reaction via Spectroscopic Ellipsometry. ACS Catal. 2020, 10, 14210–14223. [Google Scholar] [CrossRef]
- Atanasoska, L.; Atanasoski, R.; Trasatti, S. XPS and AES study of mixed layers of RuO2 and IrO2. Vacuum 1990, 40, 91–94. [Google Scholar] [CrossRef]
- Sanchez Casalongue, H.G.; Ng, M.L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. In situ observation of surface species on iridium oxide nanoparticles during the oxygen evolution reaction. Angew. Chem. 2014, 126, 7297–7300. [Google Scholar] [CrossRef]
- Liu, D.-Q.; Yu, S.-H.; Son, S.-W.; Joo, S.-K. Electrochemical performance of iridium oxide thin film for supercapacitor prepared by radio frequency magnetron sputtering method. Ecs Trans. 2008, 16, 103. [Google Scholar] [CrossRef]
- Elmaalouf, M.; Odziomek, M.; Duran, S.; Gayrard, M.; Bahri, M.; Tard, C.; Zitolo, A.; Lassalle-Kaiser, B.; Piquemal, J.-Y.; Ersen, O. The origin of the high electrochemical activity of pseudo-amorphous iridium oxides. Nat. Commun. 2021, 12, 3935. [Google Scholar] [CrossRef]
- Gao, J.; Xu, C.-Q.; Hung, S.-F.; Liu, W.; Cai, W.; Zeng, Z.; Jia, C.; Chen, H.M.; Xiao, H.; Li, J. Breaking long-range order in iridium oxide by alkali ion for efficient water oxidation. J. Am. Chem. Soc. 2019, 141, 3014–3023. [Google Scholar] [CrossRef]
- Möckl, M.; Ernst, M.F.; Kornherr, M.; Allebrod, F.; Bernt, M.; Byrknes, J.; Eickes, C.; Gebauer, C.; Moskovtseva, A.; Gasteiger, H.A. Durability testing of low-iridium PEM water electrolysis membrane electrode assemblies. J. Electrochem. Soc. 2022, 169, 064505. [Google Scholar] [CrossRef]
- Greczynski, G.; Hultman, L. C 1s peak of adventitious carbon aligns to the vacuum level: Dire consequences for material’s bonding assignment by photoelectron spectroscopy. ChemPhysChem 2017, 18, 1507–1512. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Kanaya, K.A.; Okayama, S. Penetration and energy-loss theory of electrons in solid targets. J. Phys. D Appl. Phys. 1972, 5, 43. [Google Scholar] [CrossRef]
- Seiter, J.; Müller, E.; Blank, H.; Gehrke, H.; Marko, D.; Gerthsen, D. Backscattered electron SEM imaging of cells and determination of the information depth. J. Microsc. 2014, 254, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Ferro, S.; Rosestolato, D.; Martínez-Huitle, C.A.; De Battisti, A. Charge-storage process in IrO2-SnO2 mixed-oxide electrodes. Role of coating composition, solution pH and Temperature. Electrochim. Acta 2014, 148, 85–92. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.M. Electrochemical concerted proton and electron transfers. Potential-dependent rate constant, reorganization factors, proton tunneling and isotope effects. J. Electroanal. Chem. 2006, 588, 197–206. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Chen, S.; Liang, Y.Z. Baseline correction using adaptive iteratively reweighted penalized least squares. Analyst 2010, 135, 1138–1146. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsh, P.; Huang, M.-H.; Xia, X.; Tran, I.; Atanassov, P.; Cao, H. Polarization Conforms Performance Variability in Amorphous Electrodeposited Iridium Oxide pH Sensors: A Thorough Surface Chemistry Investigation. Sensors 2024, 24, 962. https://doi.org/10.3390/s24030962
Marsh P, Huang M-H, Xia X, Tran I, Atanassov P, Cao H. Polarization Conforms Performance Variability in Amorphous Electrodeposited Iridium Oxide pH Sensors: A Thorough Surface Chemistry Investigation. Sensors. 2024; 24(3):962. https://doi.org/10.3390/s24030962
Chicago/Turabian StyleMarsh, Paul, Mao-Hsiang Huang, Xing Xia, Ich Tran, Plamen Atanassov, and Hung Cao. 2024. "Polarization Conforms Performance Variability in Amorphous Electrodeposited Iridium Oxide pH Sensors: A Thorough Surface Chemistry Investigation" Sensors 24, no. 3: 962. https://doi.org/10.3390/s24030962
APA StyleMarsh, P., Huang, M. -H., Xia, X., Tran, I., Atanassov, P., & Cao, H. (2024). Polarization Conforms Performance Variability in Amorphous Electrodeposited Iridium Oxide pH Sensors: A Thorough Surface Chemistry Investigation. Sensors, 24(3), 962. https://doi.org/10.3390/s24030962