Immobilization of DNA at Glassy Ccarbon Electrodes: A Critical Study of Adsorbed Layer

Abstract
:1. Introduction
2. Experimental
2.1. Reagents
- Probe sequence: Oligo(dG)21 3′-GGG GGG GGG GGG GGG GGG GGG-5′
- Target sequence: Oligo (dC)21 3prime;-CCC CCC CCC CCC CCC CCC CCC-5′
2.2. Apparatus
2.3. Procedure
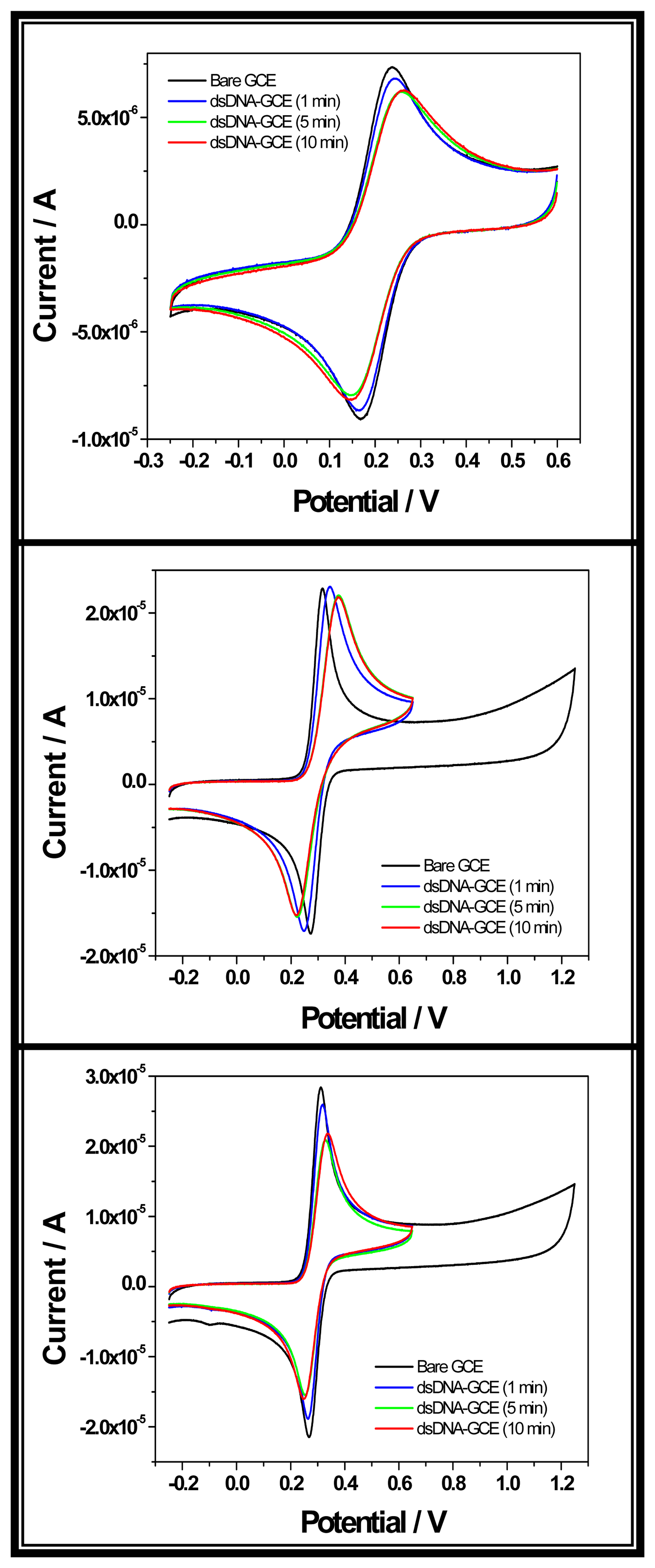
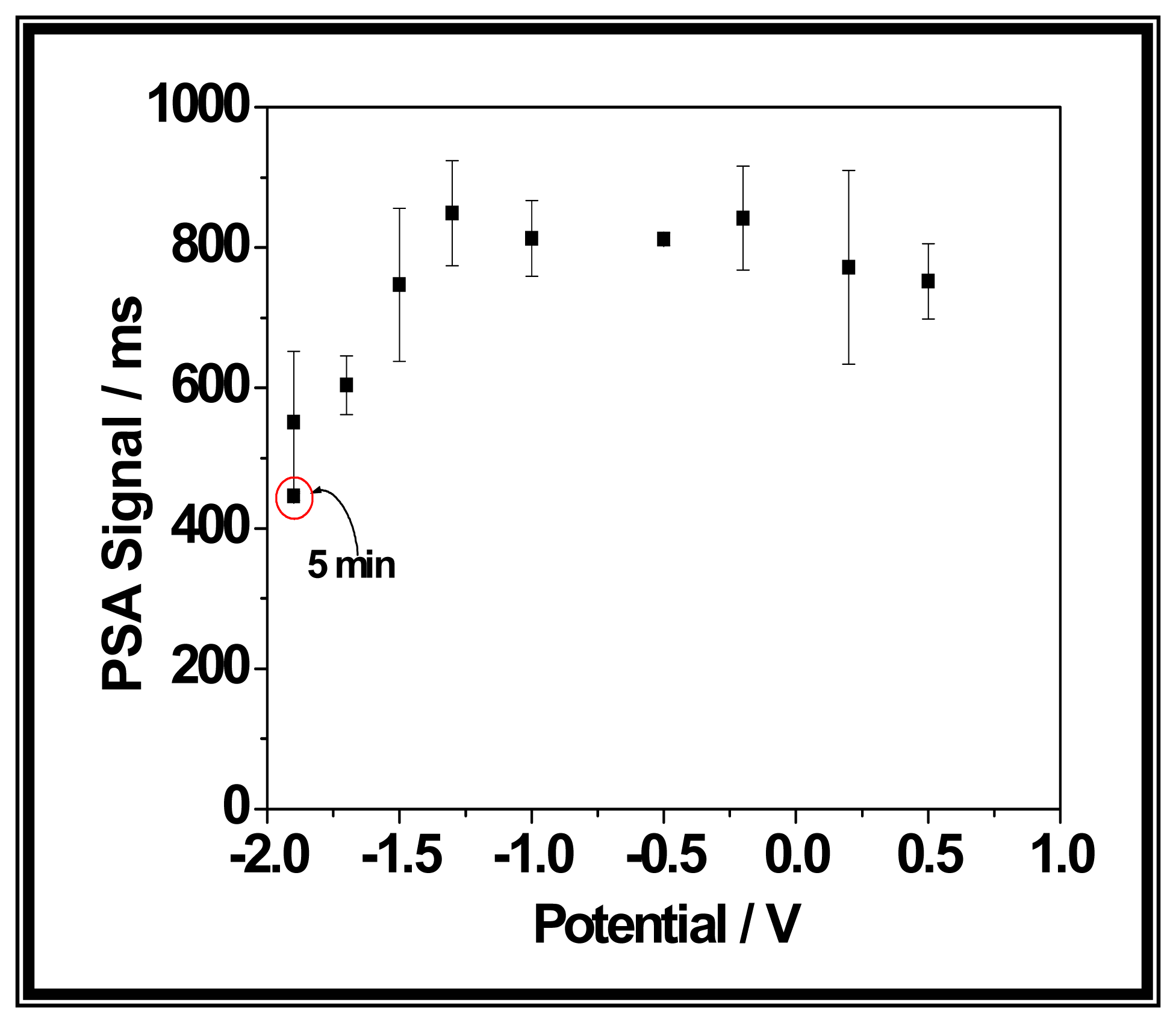
2.3.1. Electrochemical experiments
2.3.1. a. Nucleic acid adsorption and electrooxidation
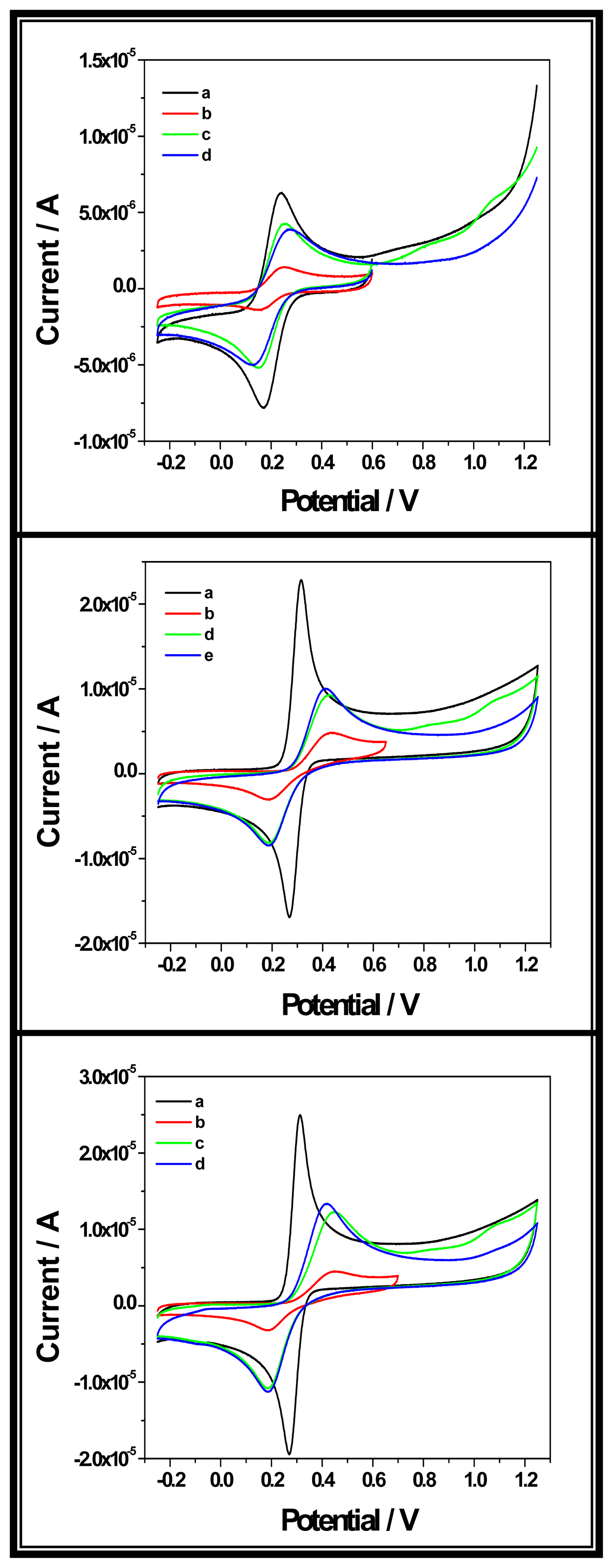
2.3.1. b. Hybridization detection
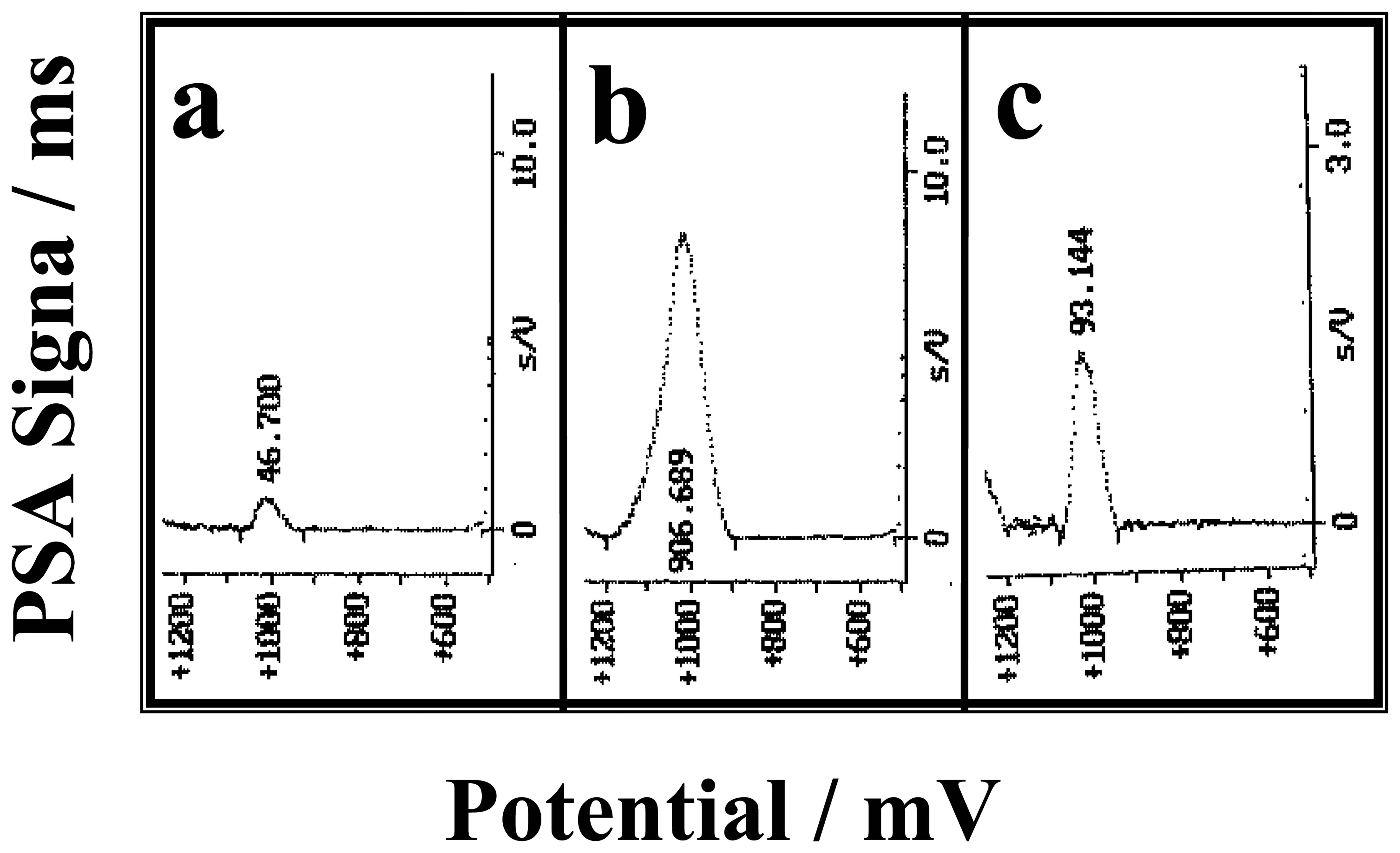
2.3.1. b.I. From the guanine oxidation signal
2.3.1. b. II. From the signal of a redox indicator
2.3.2. Raman experiments
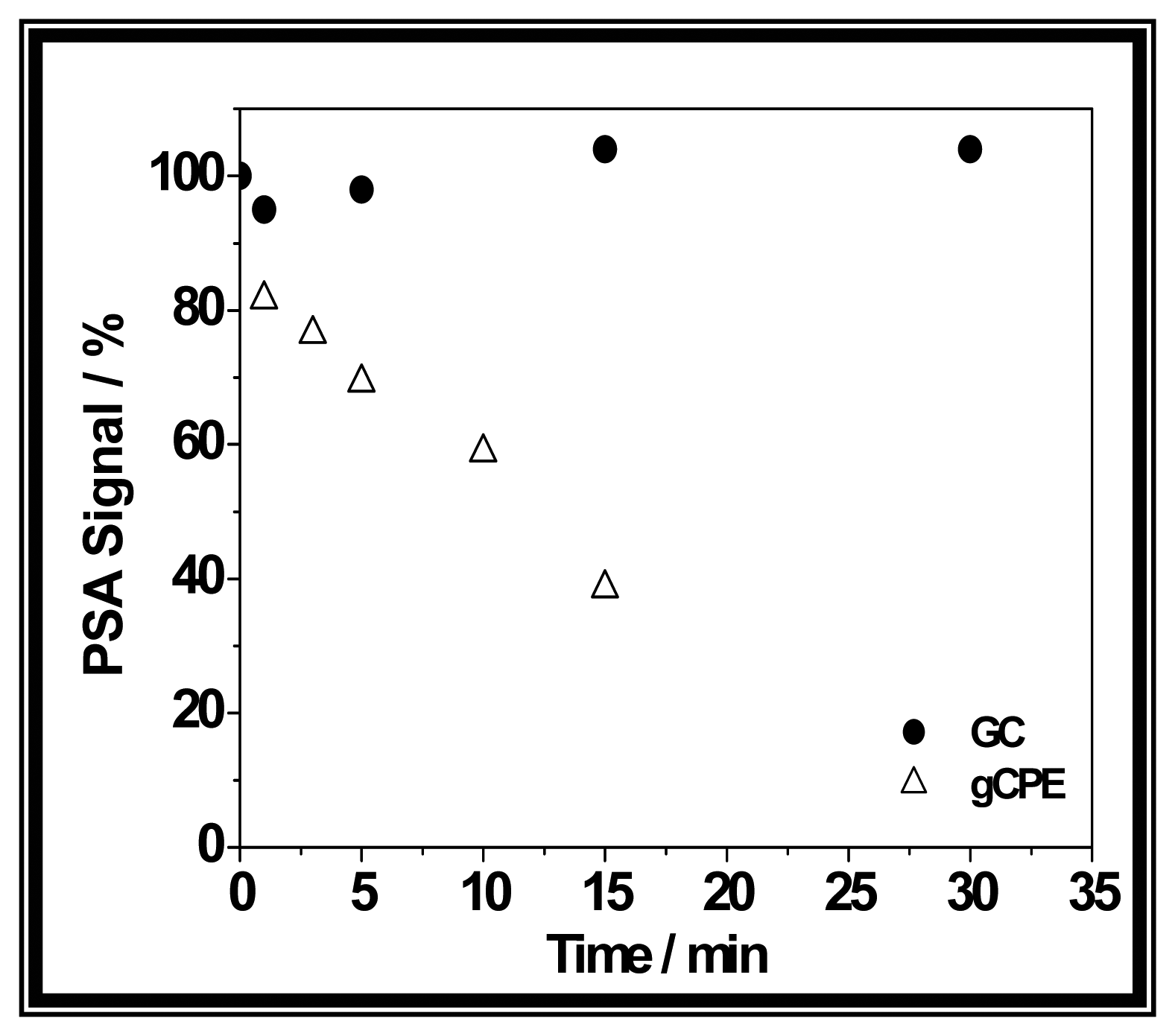
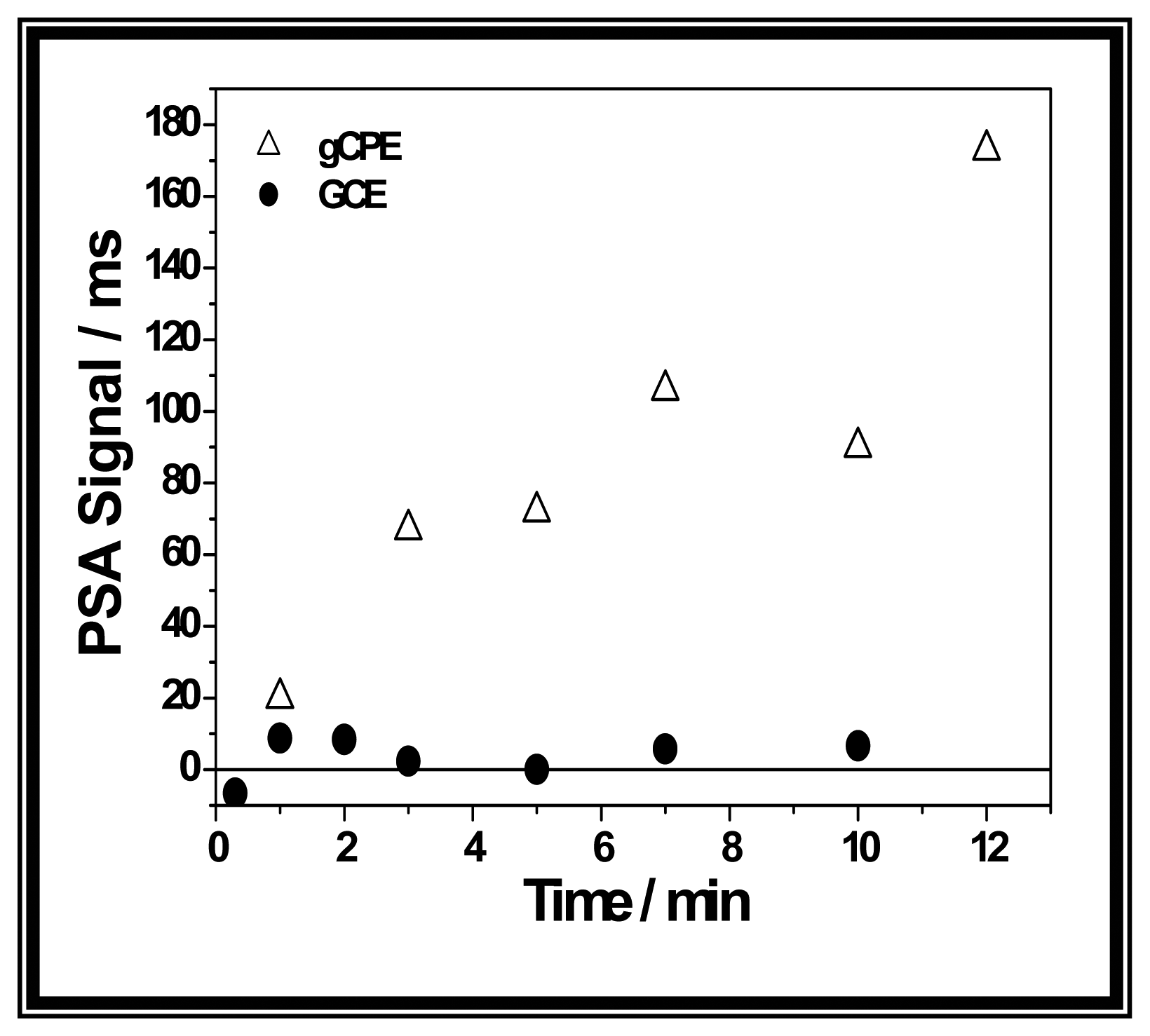
3. Results and Discussion
4. Conclusions
Acknowledgments
References and Notes
- Palecek, E. Oscillographic polarography of highly polymerized deoxyribonucleic acid. Nature. 1960, 188, 656–657. [Google Scholar]
- Wang, J. From DNA biosensors to gene chips. Nucleic Acids Research. 2000, 28, 3011–3016. [Google Scholar]
- Pividori, M.I.; Merkoci, A.; Alegret, S. Electrochemical Genosensor Design: Immobilization of Oligonucleotides onto Transducer Surfaces and Detection Methods. Biosensors and Bioelectronics. 2000, 15, 291–303. [Google Scholar]
- Palecek, E.; Fojta, M. DNA Hybridization and Damage. Anal. Chem. 2001, 73, 75A–83A. [Google Scholar]
- Palecek, E. Past, Present and Future of Nucleic Acids Electrochemistry. Talanta. 2002, 56, 809–819. [Google Scholar]
- Wang, J. Electrochemical Nucleic Acid Biosensors. Anal. Chim. Acta. 2002, 469, 63–71. [Google Scholar]
- Campas i Homs, M. DNA Sensors. Anal. Letters. 2002, 35, 1875–1894. [Google Scholar]
- Palecek, E.; Jelen, F. Electrochemistry of Nucleic Acids and Development of DNA Sensors. Critical Reviews in Anal. Chem. 2002, 32, 261–270. [Google Scholar]
- Fojta, M. Electrochemical Sensors for DNA Interactions and Damage. Electroanalysis. 2002, 14, 1449–1463. [Google Scholar]
- Erdem, A.; Ozsoz, M. Electrochemical DNA Biosensors Based on DNA-Drug Interactions. Electroanalysis 2002, 14, 965–974. [Google Scholar]
- Drummond, T.G.; Hill, M. G.; Barton, J. Electrochemical DNA Sensors. Nature Biotechnology. 2003, 21, 1192–1199. [Google Scholar]
- Lucarelli, F.; Marrazza, G.; Turner, A. P. F.; Mascini, M. Carbon and Gold Electrodes as Electrochemical Transducers for DNA Hybridisation Sensors. Biosensors and Bioelectronics. 2004, 19, 515–530. [Google Scholar]
- De-los-Santos-Álvarez, P.; Lobo-Castañón, M. J.; Miranda-Ordieres, A. J.; Tuñón Blanco, P. Electrochemistry of Nucleic Acids at Solid Electrodes and Its Applications. Electroanalysis. 2004, 16, 1193–1204. [Google Scholar]
- Wang, J.; Cai, X.; Wang, J.; Jonsson, C.; Palecek, E. Trace Measurements of RNA by Potentiometric Stripping Analysis at Carbon Paste Electrodes. Anal. Chem. 1995, 67, 4065–4070. [Google Scholar]
- Wang, J.; Cai, X.; Jonsson, C.; Balakrishnan, M. Adsorptive Stripping Potentiometry of DNA at Electrochemically Pretreated Carbon Paste Electrode. Electroanalysis. 1996, 8, 20–24. [Google Scholar]
- Cai, X.; Rivas, G.; Farias, P. A. M.; Shiraishi, H.; Wang, J.; Fojta, M.; Palecek, E. Trace Measurements of Plasmid DNAs by Adsorptive Stripping Potentiometry at Carbon Paste Electrodes. Bioelectrochem. and Bioenergetics. 1996, 40, 41–47. [Google Scholar]
- Wang, J.; Rivas, G.; Cai, X.; Chicharro, M.; Luo, D.; Palecek, E.; Nielsen, P. Adsorption and Detection of Peptide Nucleic Acids at Carbon Paste Electrodes. Electroanalysis. 1997, 9, 120–124. [Google Scholar]
- Wang, J.; Cai, X.; Fernandes, J R.; Grant, D. H.; Ozsoz, M. Electrochemical Measurements of Oligonucleotides in the Presence of Chromosomal DNA Using Membrane-Covered Carbon Electrodes. Anal. Chem. 1997, 69, 4056–4059. [Google Scholar]
- Wang, J.; Grundler, P.; Flechsing, G.-U.; Jasinski, M.; Rivas, G.; Sahlin, E.; Lopez, J. J. Stripping Analysis of Nucleic Acids at a Heated Carbon Paste Electrode. Anal. Chem. 2000, 72, 3752–3756. [Google Scholar]
- Wang, J.; Cai, X.; Tian, B.; Shiraishi, H. Microfabricated Thick-film Electrochemical Sensor for Nucleic Acid Determination. Analyst. 1996, 67, 965–969. [Google Scholar]
- Pedano, M. L.; Rivas, G. Adsorption and Electrooxidation of Carbon Nanotubes Paste Electrodes. Electrochem. Communications. 2004, 6, 10–16. [Google Scholar]
- Wang, J.; Cai, X.; Fernandes, J. R.; Grant, D. H.; Ozsoz, M. Carbon Fiber Microelectrodes for Adsorptive Stripping Analysis of Trace Nucleic Acids. J. Electroanal. Chem. 1998, 441, 167–172. [Google Scholar]
- Pedano, M. L.; Rivas, G. Immobilization of DNA on glassy carbon electrodes for the development of affinity biosensors. Biosensors and Bioelectronics. 2003, 18, 269–277. [Google Scholar]
- Wang, Z.; Liu, D.; Dong, S. Study on Adsorption and Oxidation of Calf Thymus DNA at Glassy Carbon Electrode. Electroanalysis. 2000, 12, 1419–1421. [Google Scholar]
- Wang, Z.; Liu, D.; Dong, S. In situ Infrared Spectroelectrochemical Studies on Adsorption and Oxidation of Nucleic Acids at Glassy Carbon Electrode. Bioelectrochemistry. 2001, 53, 175–181. [Google Scholar]
- Wang, H.-S.; Ju, H.-X.; Chen, H.-Y. Voltammetric Behavior and Detection of DNA at Electrochemically Pretreated Glassy Carbon Electrode. Electroanalysis. 2000, 13, 1103–1109. [Google Scholar]
- Oliveira Brett, A. M.; Serrano, S. H. P.; Gutz, I.; La-Scalea, M. A. Comparison of the Voltammetric Behavior of Metronidazole at a DNA-modified Glassy Carbon Electrode, a Mercury Thin Film Electrode and a Glassy Carbon Electrode. Electroanalysis. 1997, 9, 110–114. [Google Scholar]
- Oliveira Brett, A. M.; Serrano, S. H. P.; Gutz, I.; La-Scalea, M. A.; Cruz, M. L. Voltammetric Behavior of Nitroimidazoles at a DNA-biosensor. Electroanalysis. 1997, 9, 1132–1137. [Google Scholar]
- Oliveira Brett, A. M.; Serrano, S. H. P.; Gutz, I.; La-Scalea, M. A. Electrochemical reduction of metronidazole at a DNA-modified glassy carbon electrode. Bioelectrochem. and Bioenergetics. 1997, 42, 175–178. [Google Scholar]
- Oliveira Brett, A. M.; Macedo, T. R. A.; Raimundo, D.; Marques, M. H.; Serrano, S. H. P. Electrochemical oxidation of mitoxantrone at a glassy carbon electrode. Anal. Chim. Acta. 1999, 385, 401–408. [Google Scholar]
- Wu, K.; Fei, J.; Bai, W.; Hu, S. Direct Electrochemistry of DNA, Guanine and Adenine at a Nanostructured Film-Modified Electrode. Anal. Biochem. 2003, 376, 205–209. [Google Scholar]
- Wang, J.; Kawde, A.-N.; Sahlin, E. Renewable pencil electrodes for highly sensitive stripping potentiometric measurements of DNA and RNA. Analyst. 2000, 125, 5–7. [Google Scholar]
- Cai, X.; Rivas, G.; Farias, P. A. M.; Shiraishi, H.; Wang, J.; Palecek, E. Evaluation of Different Carbon Electrodes for Adsorptive Stripping Analysis of Nucleic Acids. Electroanalysis. 1996, 8, 753–758. [Google Scholar]
- Prado, C.; Flechsing, G.-U.; Grundler, P.; Foord, J. S.; Marken, F.; Compton, R. Electrochemical Analysis of Nucleic Acids at Boron-Doped Diamond Electrodes. Analyst. 2002, 127, 329–332. [Google Scholar]
- Brabec, V.; Dryhurst, G. Electrochemical oxidation of polyadenylic acid at graphite electrodes. J. Electroanal. Chem. 1978, 91, 219–229. [Google Scholar]
- Pang, D-W.; Zhang, M.; Wang, Z.-L.; Qi, Y.-P.; Cheng, J. K.; Liu, Z.-Y. Modification of glassy carbon and gold electrodes with DNA. J. of Electroanal. Chem. 1996, 403, 183–188. [Google Scholar]
- Wang, Z.; Liu, D.; Dong, S. Study on Adsorption and Oxidation of Calf Thymus DNA at Glassy Carbon Electrode. Electroanalysis. 2000, 12, 1419–1421. [Google Scholar]
- Oliveira Brett, A. M.; Matysik, F.- M. Voltammetric and Sonovoltammetric Studies on the Oxidation of Thymine and Cytosine at a Glassy Carbon Electrode. J. Electroanal. Chem. 1997, 429, 95–99. [Google Scholar]
- Oliveira Brett, A. M.; Piedade, J. A. P.; Silva, L. A.; Diculescu, V. C. Voltammetric Determination of all Nucleotides. Anal. Biochem. 2004, 332, 321–329. [Google Scholar]
- Wang, J.; Zhang, X.; Parrado, C.; Rivas, G. Controlled-release of DNA from Carbon-Paste Microelectrodes. Electrochem. Communications. 1999, 1, 197–202. [Google Scholar]
- DNA-modified Electrodes Part 3.: Spectroscopic Characterization of DNA-modified Gold Electrodes. Anal. Chim. Acta. 1999, 388, 93–101.
- Movileanu, L.; Benevides, J. M.; Thomas, G. J., Jr. Temperature Dependence of the Raman Spectrum of DNA. Part I - Raman Signatures of Premelting and Melting Transitions of Poly(dA-dT)·poly(dA-dT). J. Raman Spectrosc. 1999, 30, 637–649. [Google Scholar]
- Wang, J.; Cai, X.; Rivas, G.; Shiraishi, H.; Dontha, N.; Farias, P. A. M. DNA Electrochemical Biosensor for the Detection of Short DNA Sequences Related to the Human Immunodeficiency Virus. Anal. Chem. 1996, 68, 2629–2634. [Google Scholar]
- Wang, J.; Rivas, G.; Cai, X.; Dontha, N.; Shiraishi, H.; Luo, D.; Valera, F. Sequence-Specific Electrochemical Biosensing of M. tuberculosis DNA. Anal. Chim. Acta. 1997, 337, 41–48. [Google Scholar]
- Cai, X.; Rivas, G.; Shiraishi, H.; Farias, P. A. M.; Wang, J.; Tomschik, M.; Jelen, F.; Palecek, E. Electrochemical Analysis of Formation of Polynucleotide Complexes in Solution and at Electrode Surface. Anal. Chim. Acta. 1997, 344, 65–76. [Google Scholar]
- Carter, M. T.; Rodriguez, M.; Bard, A. J. Voltammetric Studies of the Interaction of Metal Chelates with DNA. 2. Tris-Chelated Complexes of Cobalt (III) and Iron (II) with 1,10-Phenanthroline and 2,2′-Bipyridine. J. Am. Chem. Soc. 1989, 111, 8901–8911. [Google Scholar]
- Pang, D.-W.; Abruña, H. D. Micromethod for the Investigation of the Interactions between DNA and Redox-Active Molecules. Anal. Chem. 1998, 70, 3162–3169. [Google Scholar]
- Labuda, J.; Bučková, M.; Vaničková, M.; Mattusch, J.; Wennrich, R. Voltammetric Detection of the DNA Interaction with Copper Complex Compounds and Damage to DNA. Electroanalysis. 1999, 11, 101–107. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dsDNA (solid) v(cm -1) | dsDNA (casting) v(cm -1) | dsDNA (potential controlled adsorption) v(cm -1) | Literature v(cm -1) | Frequency assingment |
|---|---|---|---|---|
| 720 | 760 | 750 | dT | |
| 790 | 792 | Backbone=(bk) (P-O -P) (s) | ||
| 890 | 896 | C2′H2 rk | ||
| 900 | 923 | dRibose (ring) | ||
| 1010 | 1016 | dT(CH3 ip) (rk) | ||
| 1070 | 1061 | dRib (CO) (s) | ||
| 1090 | 1092 | bk (O-P-O) (s) | ||
| 1110 | ||||
| 1145 | 1145 | 1144 | native dT | |
| 1180 | 1182 | denatured dT | ||
| 1240 | 1236 | denatured dT | ||
| 1240 | 1250 | 1256 | dA,dT | |
| 1300 | 1290 | 1301 | dA,dT | |
| 1340 | 1342 | dA | ||
| 1380 | 1376 | dT (CH3) (df),dA | ||
| 1410 | 1420 | dA,C5′H2 (df) | ||
| 1460 | 1460 | 1462 | C2′H2 (df) | |
| 1490 | 1483 | dA,dT | ||
| 1590 | 1577 | dA (N6H2) (df) | ||
| 1690 | 1673 | C4O/C5C6 (s) |
© 2005 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Pedano, M.L.; Rivas, G.A. Immobilization of DNA at Glassy Ccarbon Electrodes: A Critical Study of Adsorbed Layer. Sensors 2005, 5, 424-447. https://doi.org/10.3390/s5060424
Pedano ML, Rivas GA. Immobilization of DNA at Glassy Ccarbon Electrodes: A Critical Study of Adsorbed Layer. Sensors. 2005; 5(6):424-447. https://doi.org/10.3390/s5060424
Chicago/Turabian StylePedano, M. L., and G. A. Rivas. 2005. "Immobilization of DNA at Glassy Ccarbon Electrodes: A Critical Study of Adsorbed Layer" Sensors 5, no. 6: 424-447. https://doi.org/10.3390/s5060424
APA StylePedano, M. L., & Rivas, G. A. (2005). Immobilization of DNA at Glassy Ccarbon Electrodes: A Critical Study of Adsorbed Layer. Sensors, 5(6), 424-447. https://doi.org/10.3390/s5060424