Voltammetric Determination of Dopamine in Human Serum with Amphiphilic Chitosan Modified Glassy Carbon Electrode

Abstract
:1. Introduction
2. Experimental Section
2.1. Apparatus
2.2. Chemicals and Solutions
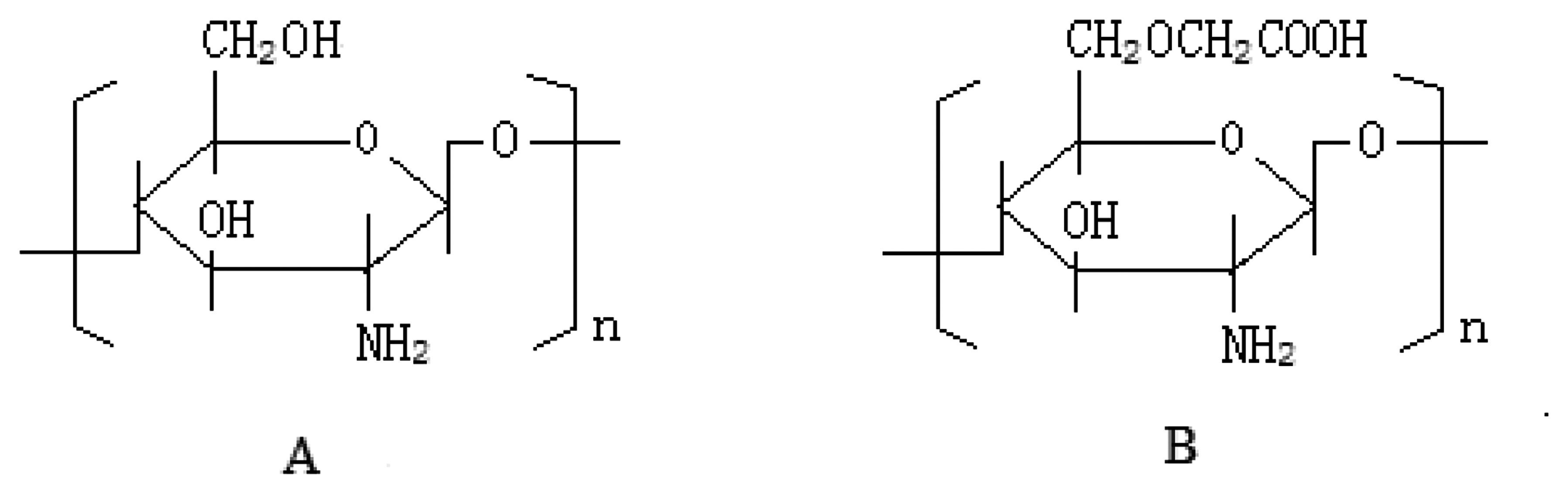
2.3. OCMCS Synthesis [12]
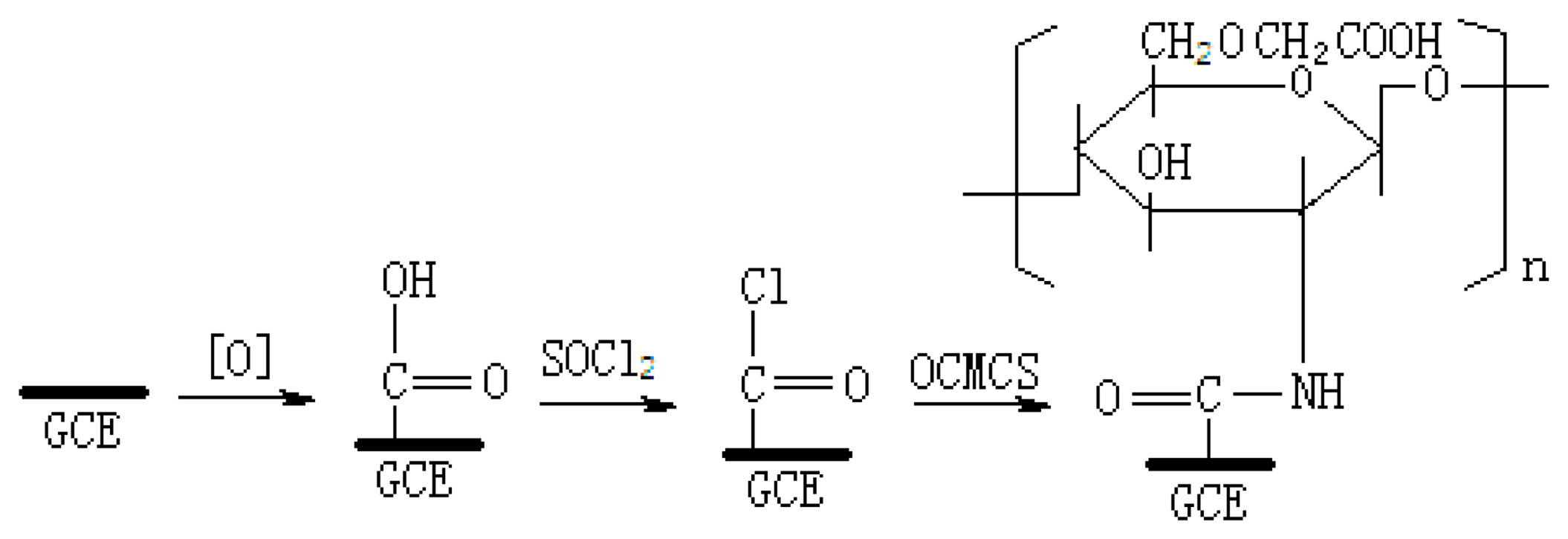
2.4. Fabrication of the OCMCS-modified Glassy Carbon Electrodes
2.5. Determination of DA
3. Results and Discussion
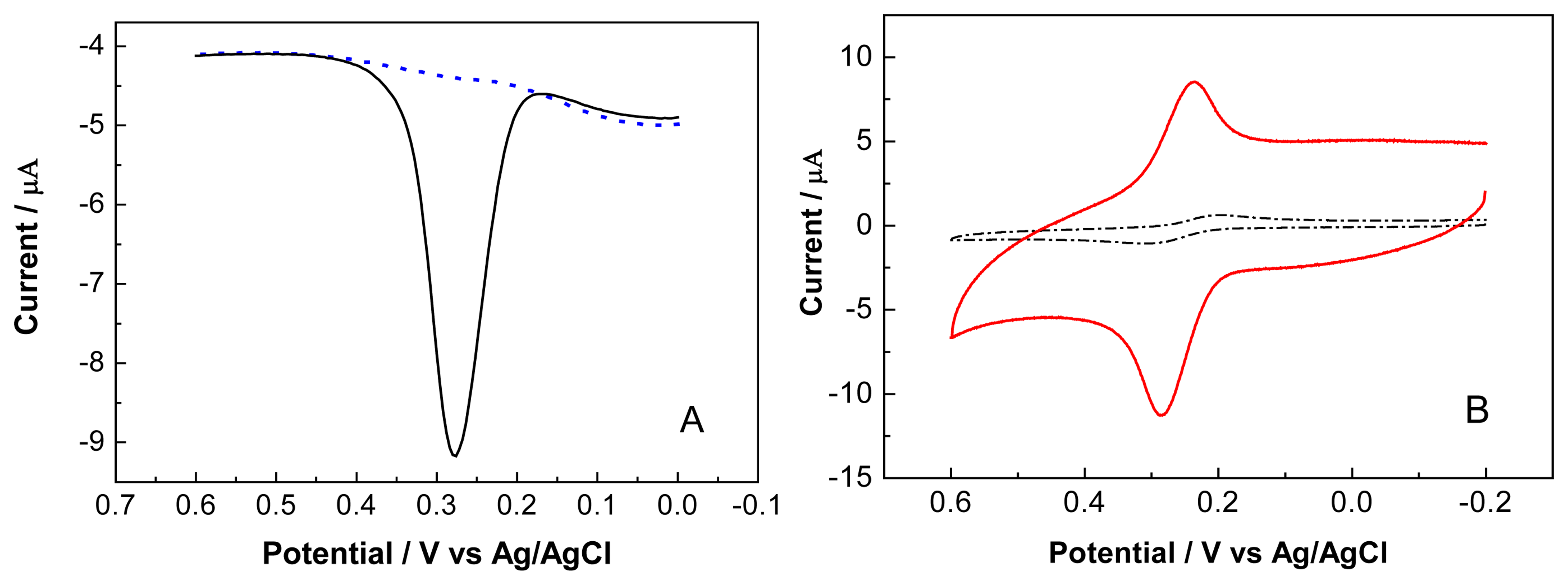
3.1. The Role of the OCMCS at the Modified Electrode
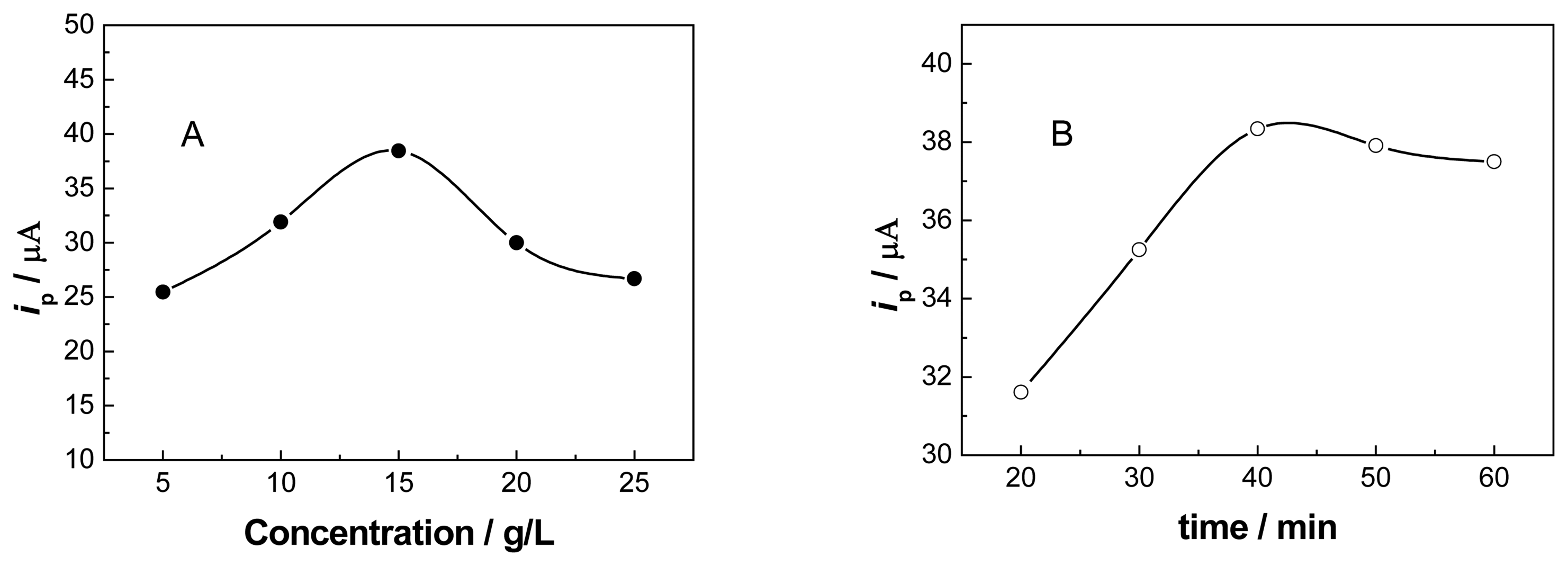
3.2. Optimization of the GCE Modification Conditions
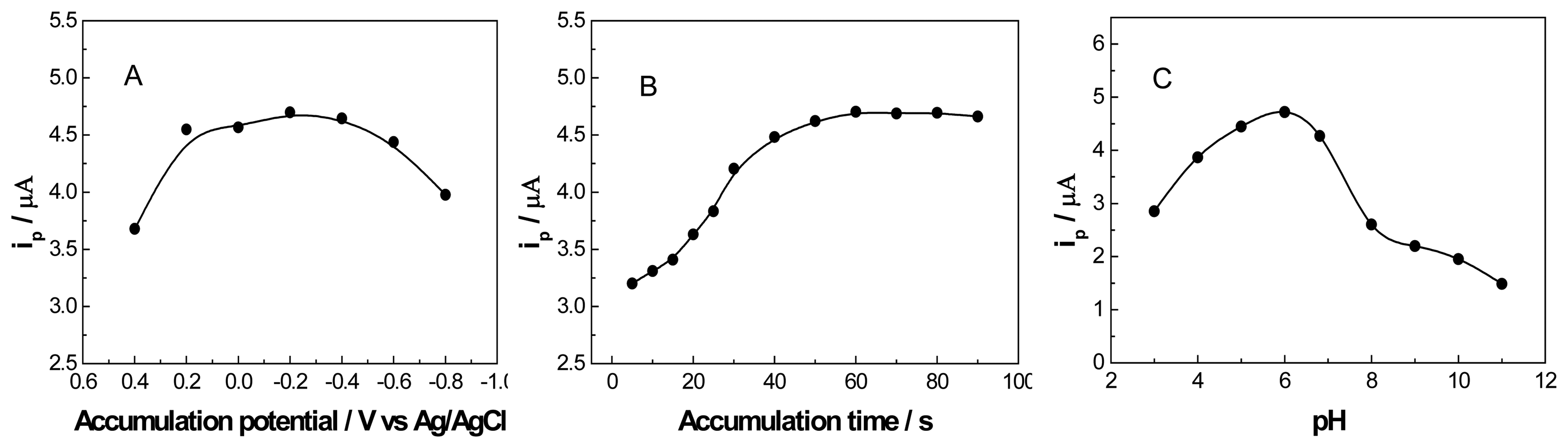
3.3. Optimization of Experimental Conditions of the Determination
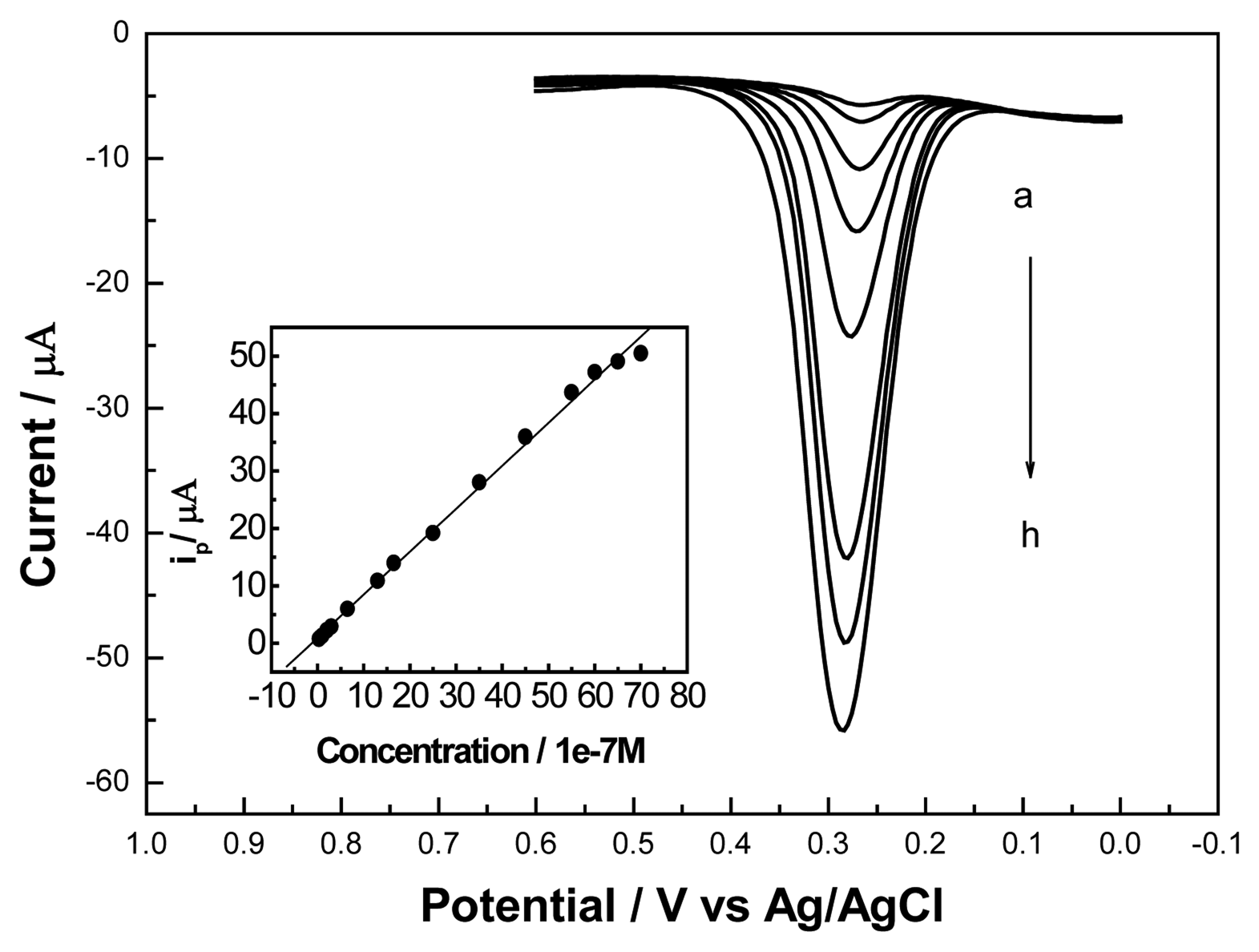
3.4. Calibration Curve and Detection Limit
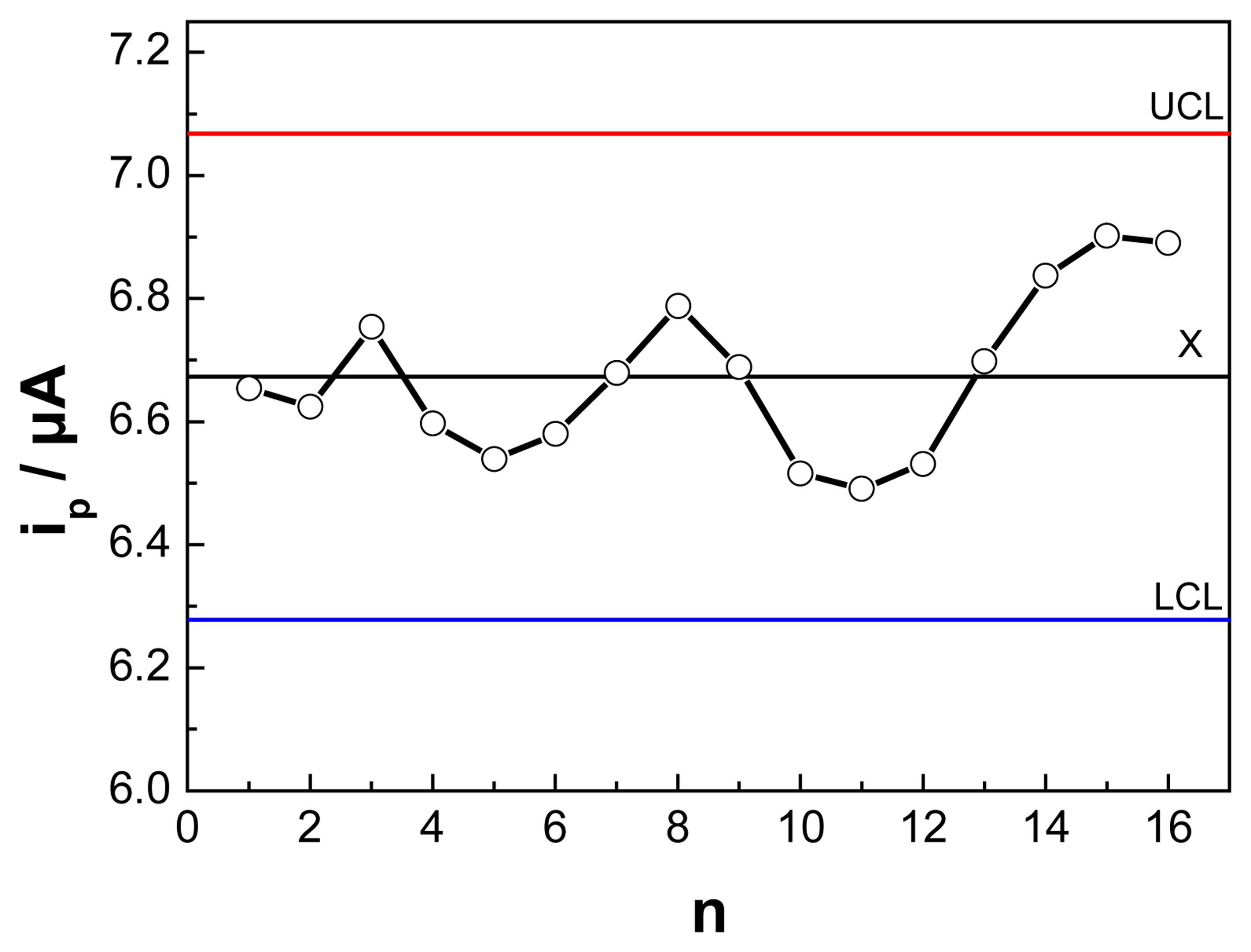
3.5. Reproducibility and Lifetime of the OCMCS/GCE
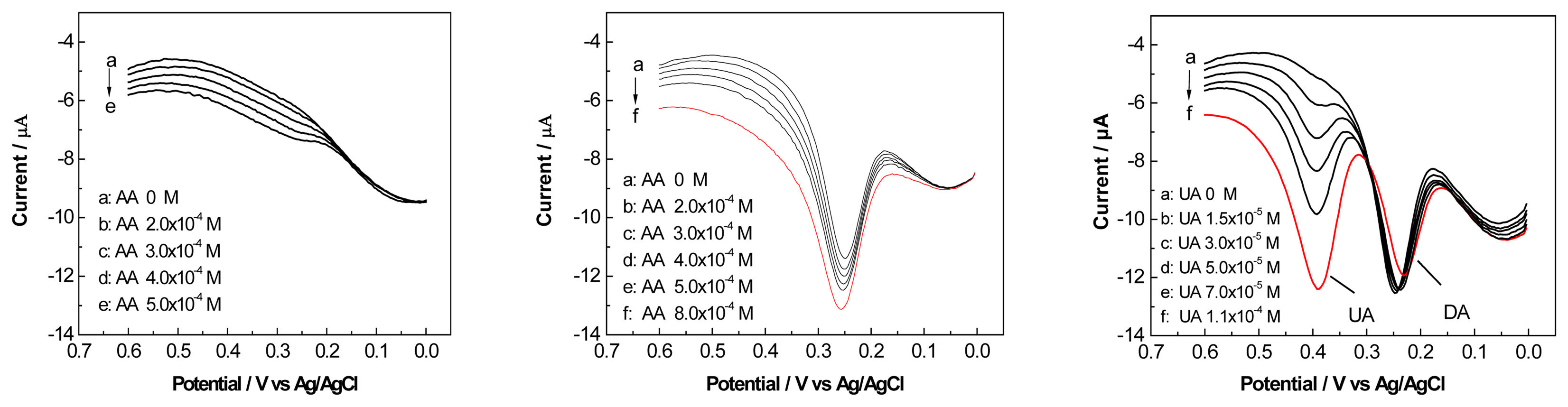
3.6. Interference
3.7. Detection of DA in the Human Serum Sample
4. Conclusions
Acknowledgments
References
- Osuji, G. O.; Madu, W. C. Ammonium ion salvage by glutamate dehydrogenase during defence response in maize. Phytochem. 1996, 42, 1491–1498. [Google Scholar]
- Yu, H.; Yan, F.; Dai, Z.; Ju, H. X. A disposable amperometric immunosensor for α-1-fetoprotein based on enzyme-labled antibody/chitosan-membrane-modified screen-printed carbon electrode. Anal. Biochem. 2004, 331, 98–105. [Google Scholar]
- Lei, C. X.; Gong, F. C.; Shen, G. L.; Yu, R. Q. Amperometric immunosensor for Schistosoma japonicum antigen using antibodies loaded on a nano-Au monolayer modified chitosan-antrapped carbon paste electrode. Sensor. Actuat. B 2003, 96, 582–588. [Google Scholar]
- Yang, M. H.; Yang, Y. H.; Liu, B.; Shen, G. L.; Yu, R. Q. Amperometric glucose biosensor based on chitosan with improved selectivity and stability. Sensor. Actuat. B 2004, 101, 269–276. [Google Scholar]
- Xu, J. J.; Luo, X. L.; Du, Y.; Chen, H. Y. Application of MnO2 nanoparticles as an eliminator of ascorbate interference to amperometric glucose biosensors. Electrochem. Commun. 2004, 6, 1169–1173. [Google Scholar]
- Miao, Y.; Tan, S. N. Amperometric hydrogen peroxide biosensor with silica sol-gel/chitosan film as immobilization matrix. Anal. Chim. Acta 2001, 437, 87–93. [Google Scholar]
- Wang, G.; Xu, J. J.; Chen, H. Y.; Lu, Z. H. Amperometric hydrogen peroxide biosensor with sol-gel/chitosan network-like film as immobilization matrix. Biosens. Bioelectron. 2003, 18, 335–343. [Google Scholar]
- Magalhaes, J. M. C. S.; Machado, A. A. S. C. Urea potentiometric biosensor based on urease immobilized on chitosan membranes. Talanta 1998, 47, 183–191. [Google Scholar]
- Xu, J. R.; Lin, B. Preconcentration and determination of lead ions at a chitosan-modified glassy carbon electrode. Analyst 1994, 119, 1599–1601. [Google Scholar]
- Zhao, C. Z.; Pan, Y. Z.; Su, Y.; Zhang, Z. H.; Guo, Z.; Sun, L. C. Determination of EDTA species in water by square-wave voltammetry using a chitosan-coated glassy carbon electrode. Water Res. 2003, 37, 4270–4274. [Google Scholar]
- Lu, G. H.; Yao, X.; Wu, A. G.; Zhan, T. Determination of the total iron by chitosan-modified glassy carbon electrode. Microchem. J. 2001, 69, 81–87. [Google Scholar]
- Zhu, A. P.; Chan-Park, B. M. B.; Dai, S.; Li, L. The aggregation behavior of O-carboxymethylchitoan in dilute aqueous solution. Colloid. Surf. B 2005, 43, 143–149. [Google Scholar]
- Zhao, H.; Zhang, Y. Z.; Yuan, Z. B. Electrochemical determination of dopamine using a poly(2-picolinic acid) modified glassy carbon electrode. Analyst 2001, 126, 358–360. [Google Scholar]
- Li, J.; Lu, J. Flow-injection/chemiluminescene assays of catecholamines. Chinese J. Anal. Chem. 1997, 25, 314–316. [Google Scholar]
- Nohta, H.; Yukizawa, T.; Ohkura, Y.; Yoshimura, M.; Ishida, J.; Yamaguchi, M. Aromatic glycinonitriles and methylamines as pre-column fluorescene derivatization reagents for catecholamines. Anal. Chim. Acta 1997, 344, 233–240. [Google Scholar]
- Wu, Y.; Fan, R.; Di, J. Electrochemical study of electron transfer between dopamine and ferrocene at liquid/liquid interface. Chinese J. Anal. Chem. 1996, 24, 873–875. [Google Scholar]
- Zhu, R.; Kok, W. T. Determination of catecholamines and related compounds by capillary electrophoresis with postcolumn terbium complexation and sensitized luminescence detection. Anal. Chem. 1997, 69, 4010–4016. [Google Scholar]
- Xiao, L. F.; Chen, J.; Cha, C. S. Elimination of the interference of ascorbic acid in the amperometric detection of biomolecules in body fluid samples and the simple detection of uric acid in human serum and urine by using the powder microelectrode technique. J. Electroanal. Chem. 2000, 495, 27–35. [Google Scholar]
- Zeng, Y. L.; Li, C. X.; Tang, C. R.; Zhang, X. B.; Shen, G. L.; Yu, R. Q. The electrochemical properties of Co (TPP), tetraphenylborate modified glassy carbon electrode: application to dopamine and uric acid analysis. Electroanal. 2006, 18, 440–448. [Google Scholar]
- Zhao, H.; Zhang, Y. Z.; Yuan, Z. B. Electrochemical determination of dopamine using a poly (2-picolinic acid) modified glassy electrode. Analyst 2001, 126, 358–360. [Google Scholar]
- Downard, A. J.; Roddick, A. D.; Bond, A. M. Covalent modification of carbon electrode for voltammetric differentiation of dopamine and ascorbic acid. Anal. Chim. Acta 1995, 317, 303–310. [Google Scholar]
- Zhao, H.; Zhang, Y. Z.; Yuan, Z. B. Study on the electrochemical behavior of dopamine with poly (sulfosalicyclic acid) modified glassy carbon electrode. Anal. Chim. Acta 2001, 441, 117–122. [Google Scholar]
- Ciszewski, A.; Milczarek, G. Polyeugenol-modified platinum electrode for selective detection of dopamine in the presence of ascorbic acid. Anal. Chem 1999, 71, 1055–1061. [Google Scholar]
- Kang, T. F.; Shen, G. L.; Yu, R. Q. Voltammetric behavior of dopamine at nickel phthalocyanine polymer modified electrodes and analytical applications. Anal. Chim. Acta 1997, 356, 245–251. [Google Scholar]
- Zheng, L. Z.; Wu, S. G.; Lin, X. Q.; Nie, L.; Rui, L. Selective determination of dopamine in the presence of ascorbic acid at an over-oxidized poly (N-acetylanine) electrode. Analyst 2001, 126, 736–738. [Google Scholar]
- Kang, T. F.; Shen, G. L.; Yu, R. Q. Permselectivity of neurotransmitters at overoxidized polypyrrole film coated glassy carbon electrodes. Talanta 1996, 43, 2007–2013. [Google Scholar]
- Pihel, K. R.; Walker, Q. D.; Wightman, R. M. Overoxidized polypyrrole-coated carbon fiber microelectrodes for dopamine measurements with fast-scan cyclic voltammetry. Anal. Chem. 1996, 68, 2084–2089. [Google Scholar]
- Zen, J. M.; Wang, W. M.; Ilangovan, G. Adsorptive potentiometric stripping analysis of dopamine on clay/GCE. Anal. Chim. Acta 1998, 372, 315–321. [Google Scholar]
- Hocevar, S. B.; Wang, J.; Deo, R. P.; Musameh, M.; Ogorevc, B. Carbon nanotube modified microelectrode for enhanced voltammetric detection of dopamine in the presence of ascorbate. Electroanal. 2005, 17, 417–422. [Google Scholar]
- Valentini, F.; Orlanducci, S.; Tamburri, E.; Terranova, M. L.; Curulli, A.; Palleschi, G. Single-walled carbon nanotubes on tungsten wires: a new class of microelectrochemical sensors. Electroanal. 2005, 17, 28–37. [Google Scholar]
- Chen, R. S.; Huang, W. H.; Tong, H.; Wang, Z. L.; Cheng, J. K. Carbon fiber nanoelectrodes modified by single-walled carbon nanotubes. Anal. Chem. 2003, 75, 6341–6345. [Google Scholar]
- Wang, Z. H.; Liang, Q. L.; Wang, Y. M.; Luo, G. A. Carbon nanotube-intercalated graphite electrodes for simultaneous determination of dopamine and serotonin in the presence of ascorbic acid. J. Electroanal. Chem. 2003, 540, 129–134. [Google Scholar]
- Wang, Q.; Jiang, N.; Li, N. Q. Electrocatalytic response of dopamine at a thiolactic acid self-assembled gold electrode. Microchem. J. 2001, 68, 77–85. [Google Scholar]
- Zhang, H. M.; Li, N. Q.; Zhu, Z. W. Electrocatalytic response of dopamine at a DL-homocysteine self-assembled gold electrode. Microchem. J. 2000, 64, 277–282. [Google Scholar]
- Liu, T.; Li, M. X.; Li, Q. Y. Electroanalysis of dopamine at a gold electrode modified with N-acetylcysteine self-assembled monolayer. Talanta 2004, 63, 1053–1059. [Google Scholar]
- Wang, Q.; Dong, D.; Li, N. Q. Electrochemical response of dopamine at a penicillamine self-assembled gold electrode. Bioelectrochem. 2001, 54, 169–175. [Google Scholar]
- Raj, C. R.; Okajima, T.; Ohsaka, T. Gold nanoparticle arrays for the voltammetric sensing of dopamine. J. Electroanal. Chem. 2003, 543, 127–133. [Google Scholar]
- Zhang, L.; Jia, J. B.; Zou, X. Q.; Dong, S. J. Simultaneous determination of dopamine and ascorbic acid at an in-site functionalized self-assembled monolayer on gold electrode. Electroanal. 2004, 16, 1413–1418. [Google Scholar]
- Zare, H. R.; Nasirizadeh, N.; Ardakani, M. M. Electrochemical properties of a tetrabromo-p-benzoquinone modified carbon paste electrode. Application to the simultaneous determination of ascorbic acid, dopamine and uric acid. J. Electroanal. Chem. 2005, 577, 25–33. [Google Scholar]
- Zhao, Y. F.; Gao, Y. Q.; Zhan, D. P.; Liu, H. Selective detection of dopamine in the presence of ascorbic acid and uric acid by a carbon nanotubes-ionic liquid gel modified electrode. Talanta 2005, 66, 51–57. [Google Scholar]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar]
- Ramesh, P.; Sampath, S. Selective determination of uric acid in presence of ascorbic acid and dopamine at neutral pH using exfoliated graphite electrodes. Electroanal. 2004, 16, 866–869. [Google Scholar]
- Wang, C. Y.; Liu, Q. X.; Shao, X. Q.; Xue, H. G.; Hu, X. Y. One step fabrication of nanoelectrode ensembles formed via amphiphilic block copolymers self-assembly and selective voltammetric detection of uric acid in the presence of high ascorbic acid content. Talanta 2006, in press. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interferent | Concentration / mM | Signal change % (iDA= 100 %) |
|---|---|---|
| AA | 0.30 | + 2.4 |
| 0.40 | + 3.0 | |
| 0.50 | + 3.5 | |
| UA | 0.03 | - 1.8 |
| 0.05 | - 2.4 | |
| 0.07 | - 3.1 | |
| urea | 0.50 | + 2.7 |
| tartaric acid | 0.05 | - 0.5 |
| D-fructose | 0.50 | - 2.8 |
| citrate | 0.05 | + 2.1 |
| glucose | 0.50 | - 0.7 |
| cysteine | 0.05 | - 2.9 |
| DL-tyrosine | 0.05 | - 2.3 |
| L-alanine | 0.50 | - 0.5 |
| caffeine | 0.05 | + 1.5 |
| vitamine B1 | 0.50 | + 2.2 |
| vitamine B2 | 0.50 | + 2.7 |
| vitamine B6 | 0.50 | + 2.0 |
| NaNO3 | 0.50 | - 3.0 |
| (NH4)2SO4 | 0.50 | - 2.3 |
| KCl | 0.50 | - 1.9 |
| MgCl2 | 0.05 | - 2.9 |
| BaCl2 | 0.05 | - 3.0 |
| Fe(NO3)3 | 0.05 | - 3.4 |
| Serum | Spiked / μg | Detected / μg | Recovery / % |
|---|---|---|---|
| Sample 1 | 0.95 | 0.92 | 96.8 |
| Sample 2 | 1.71 | 1.71 | 100.0 |
| Sample 3 | 4.55 | 4.54 | 99.8 |
| Sample 4 | 9.48 | 9.79 | 103.3 |
| Sample 5 | 13.39 | 13.50 | 100.8 |
| Sample 6 | 18.02 | 18.15 | 100.7 |
© 2006 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Wang, C.Y.; Wang, Z.X.; Zhu, A.P.; Hu, X.Y. Voltammetric Determination of Dopamine in Human Serum with Amphiphilic Chitosan Modified Glassy Carbon Electrode. Sensors 2006, 6, 1523-1536. https://doi.org/10.3390/s6111523
Wang CY, Wang ZX, Zhu AP, Hu XY. Voltammetric Determination of Dopamine in Human Serum with Amphiphilic Chitosan Modified Glassy Carbon Electrode. Sensors. 2006; 6(11):1523-1536. https://doi.org/10.3390/s6111523
Chicago/Turabian StyleWang, Cheng Yin, Zhi Xian Wang, Ai Ping Zhu, and Xiao Ya Hu. 2006. "Voltammetric Determination of Dopamine in Human Serum with Amphiphilic Chitosan Modified Glassy Carbon Electrode" Sensors 6, no. 11: 1523-1536. https://doi.org/10.3390/s6111523
APA StyleWang, C. Y., Wang, Z. X., Zhu, A. P., & Hu, X. Y. (2006). Voltammetric Determination of Dopamine in Human Serum with Amphiphilic Chitosan Modified Glassy Carbon Electrode. Sensors, 6(11), 1523-1536. https://doi.org/10.3390/s6111523