Modelling the Transport and Kinetics of Electroenzymes at the Electrode/Solution Interface

Abstract
:Introduction

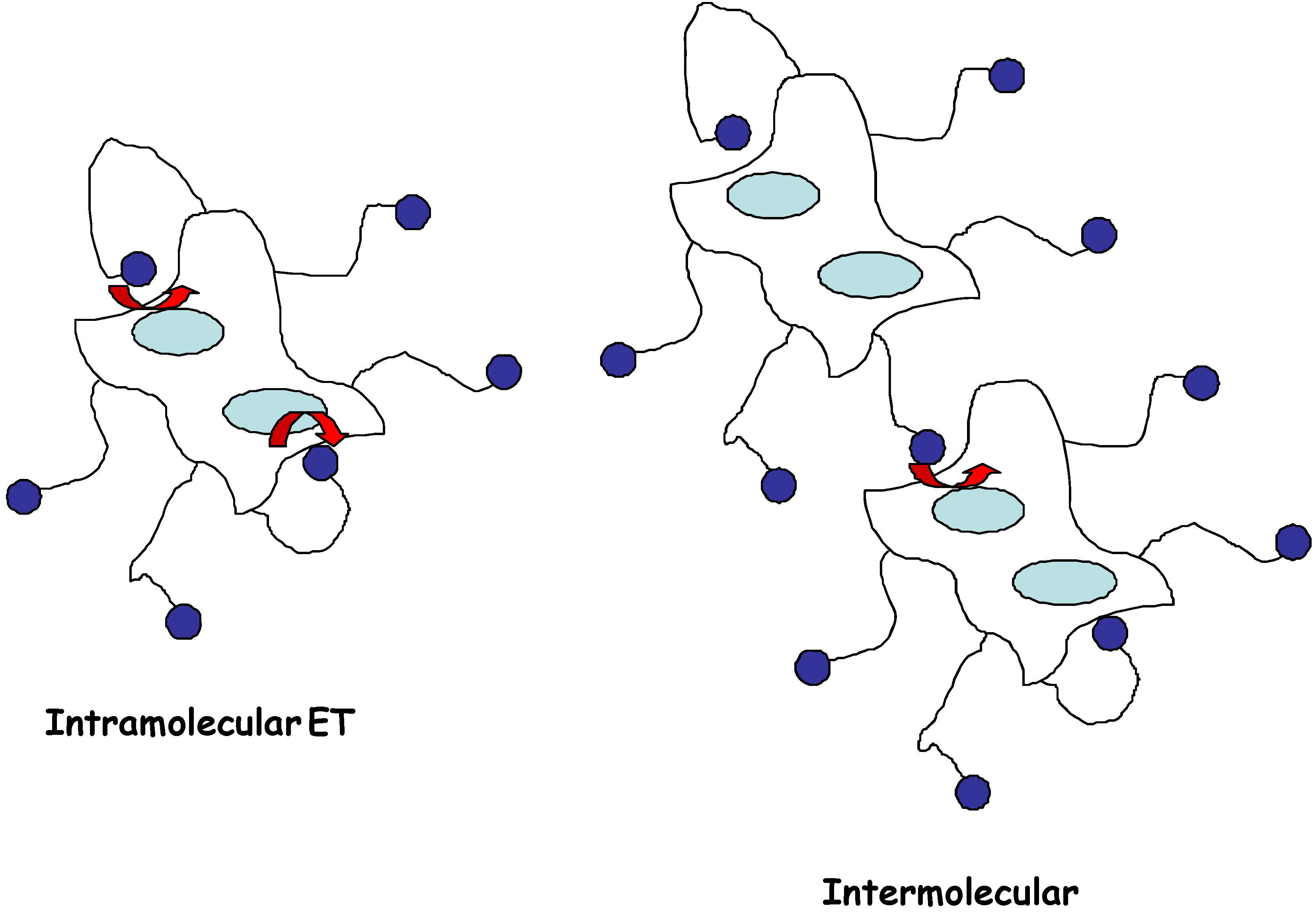
Direct reaction of redox enzymes at electrodes
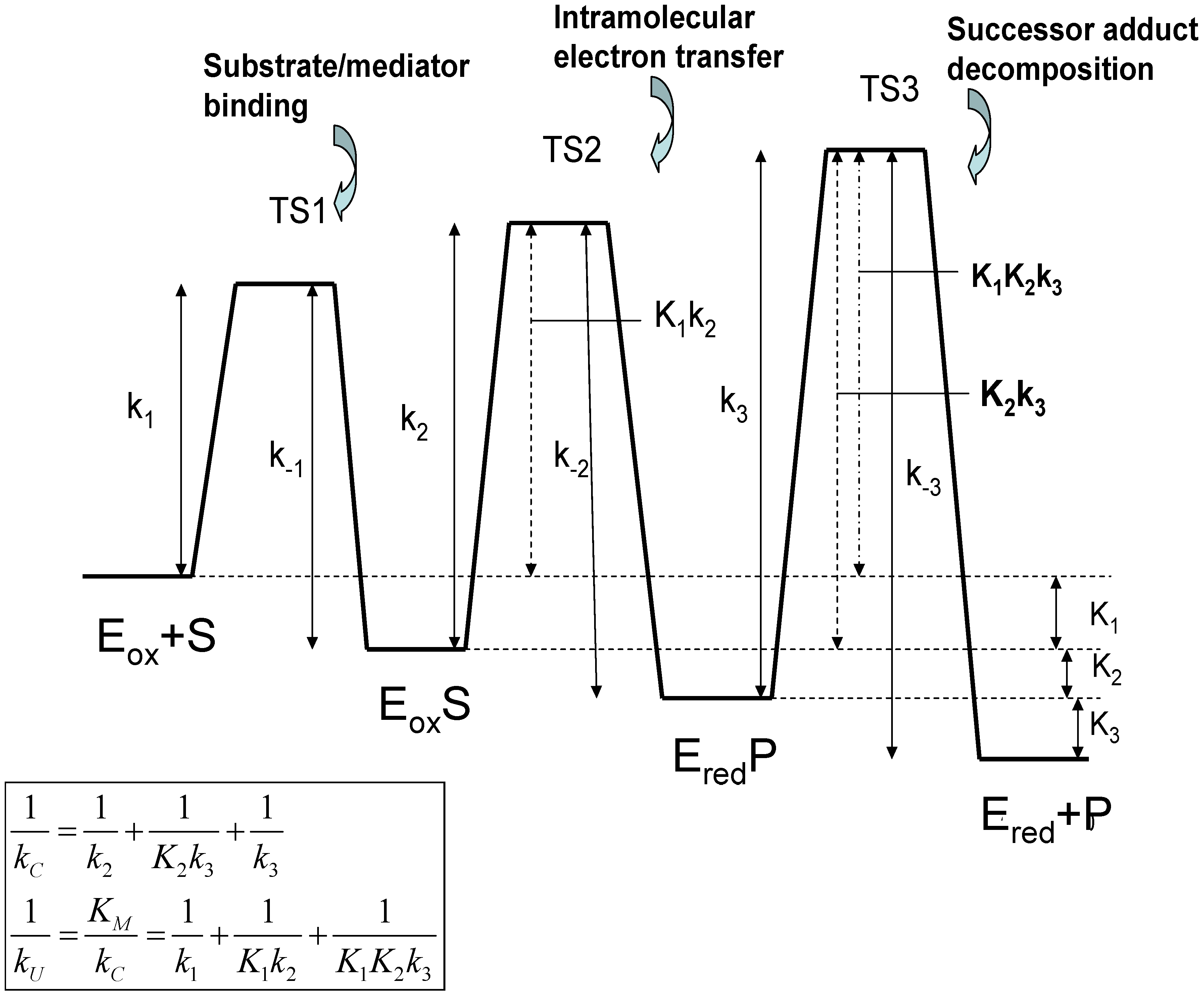
Development of the mathematical model

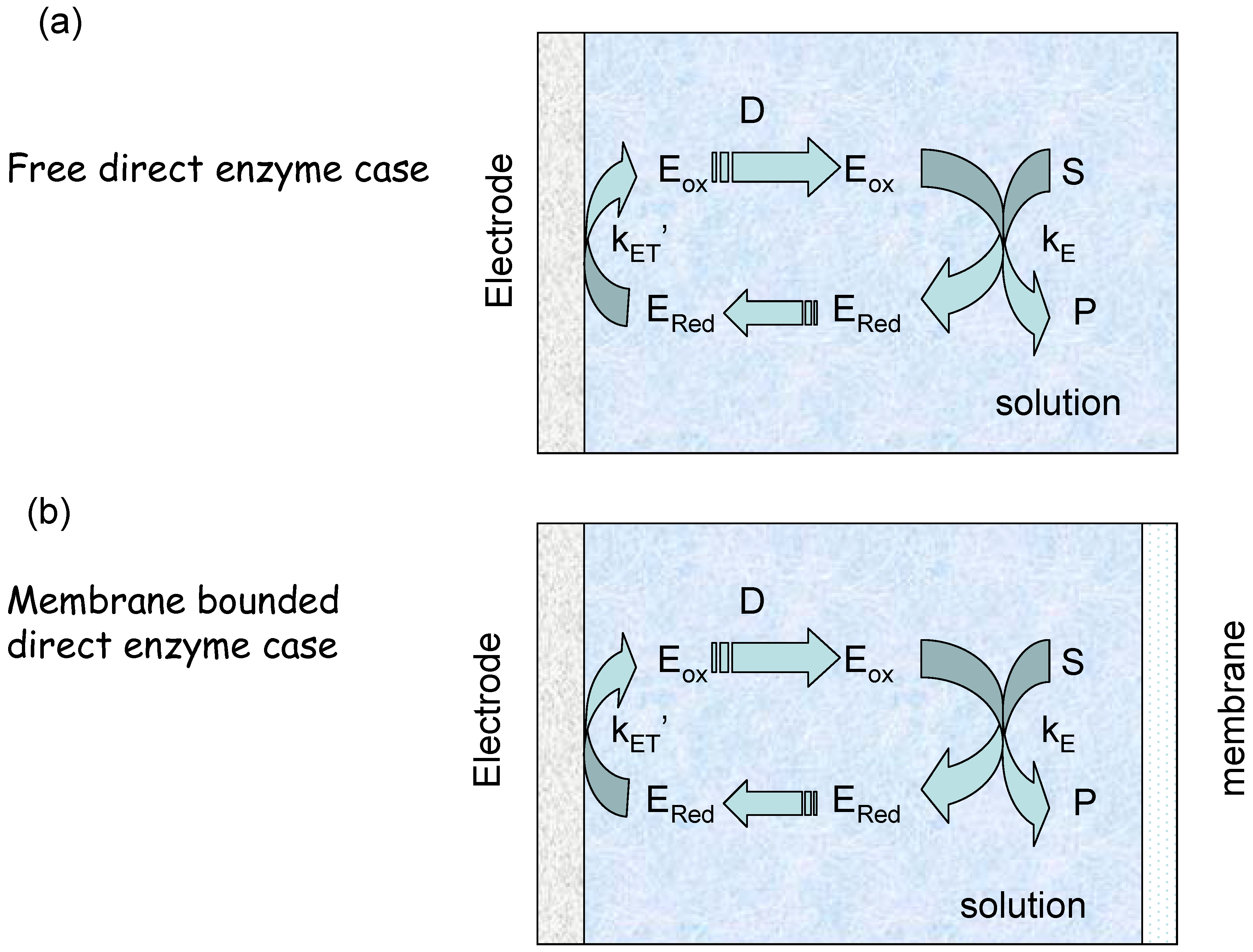
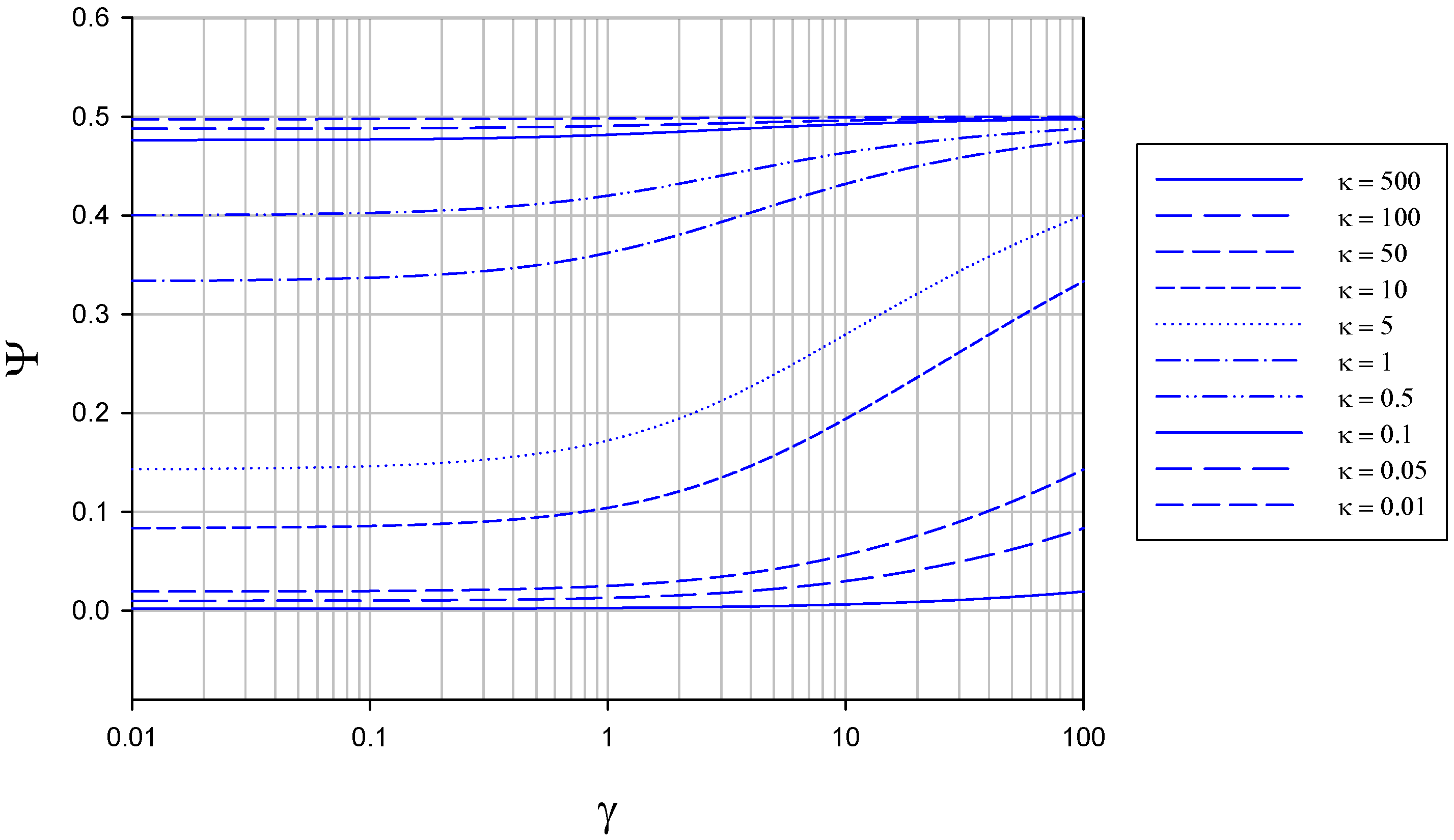
The membrane free situation



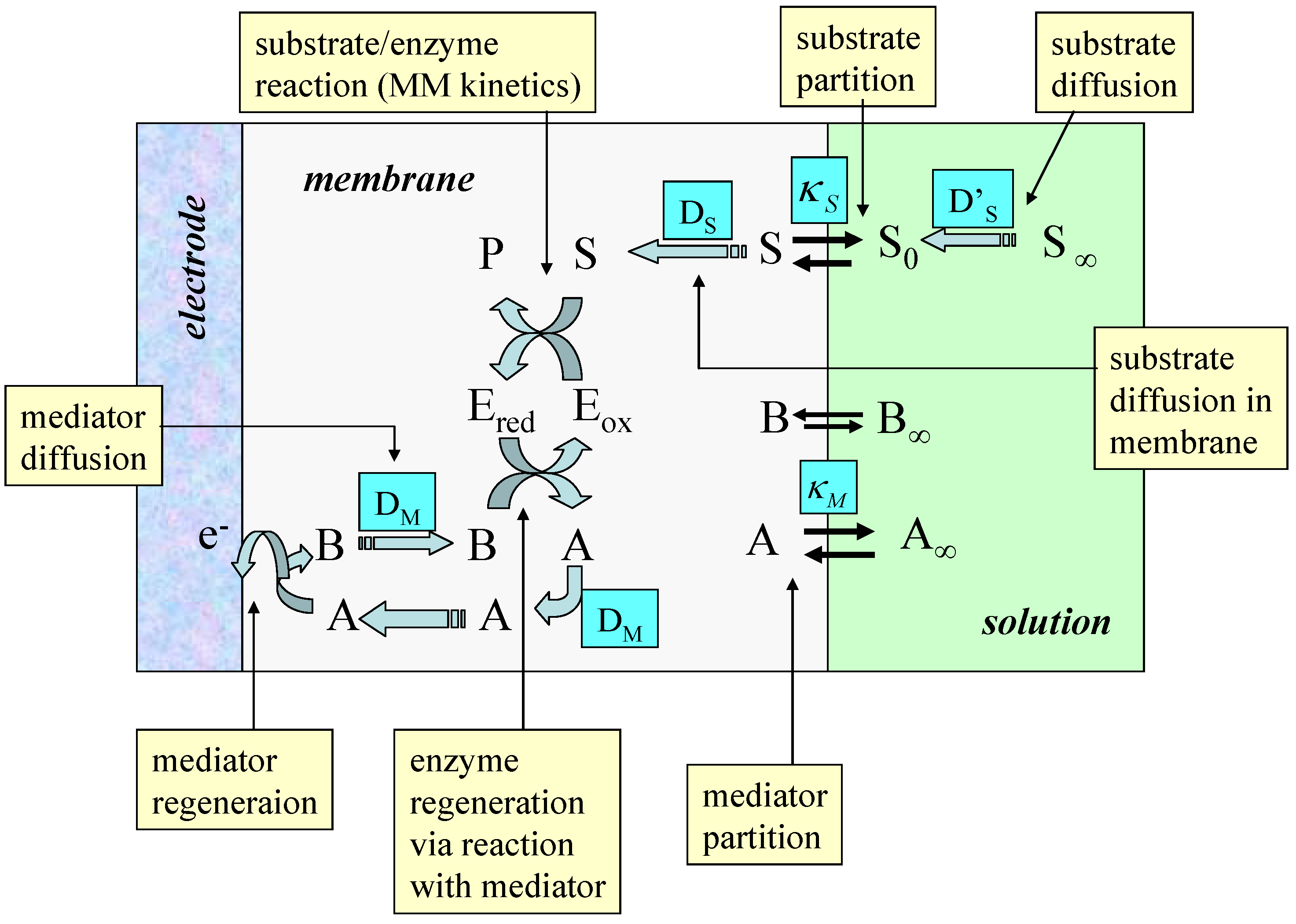
The membrane bounded situation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
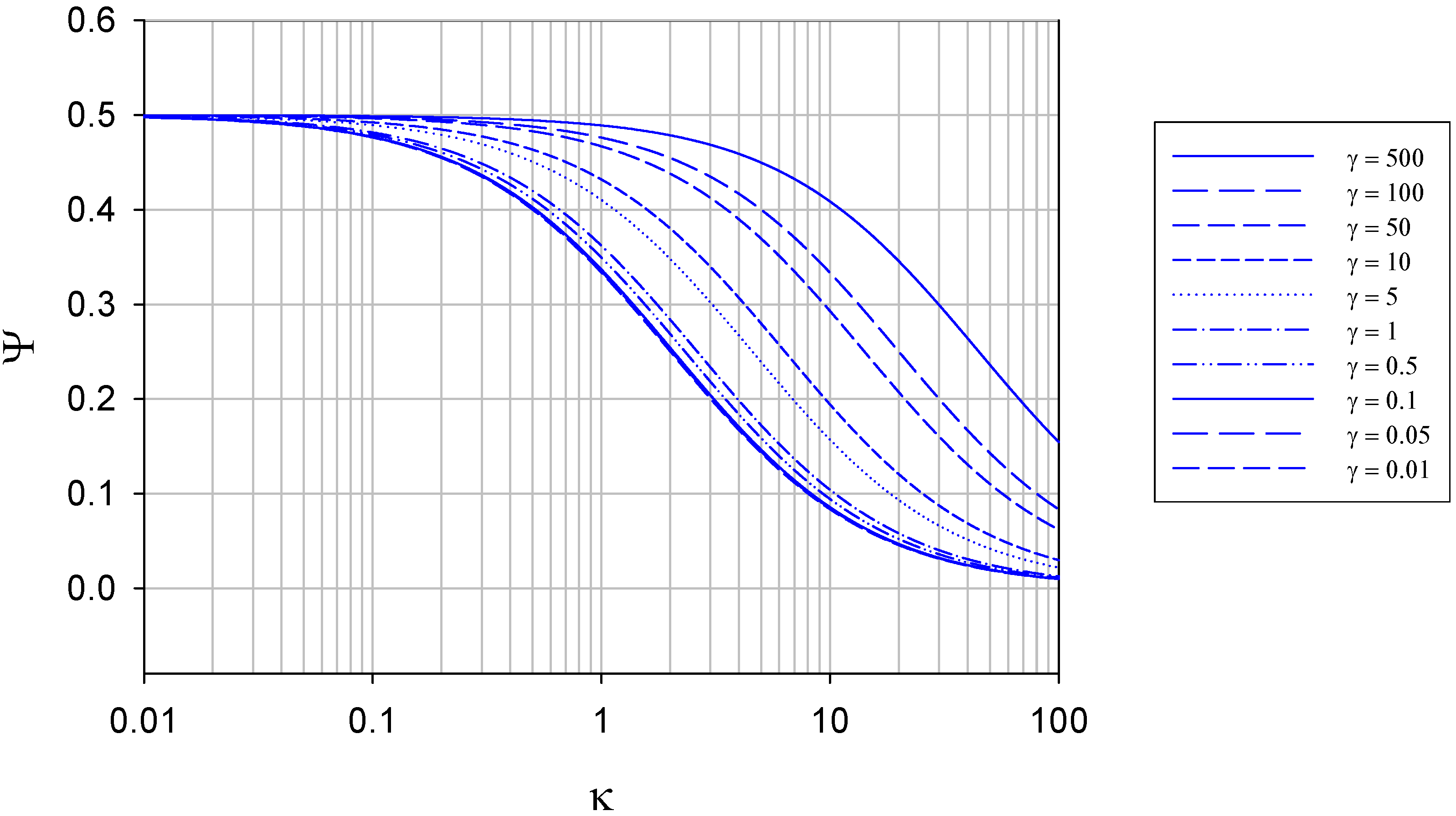{kind=link}
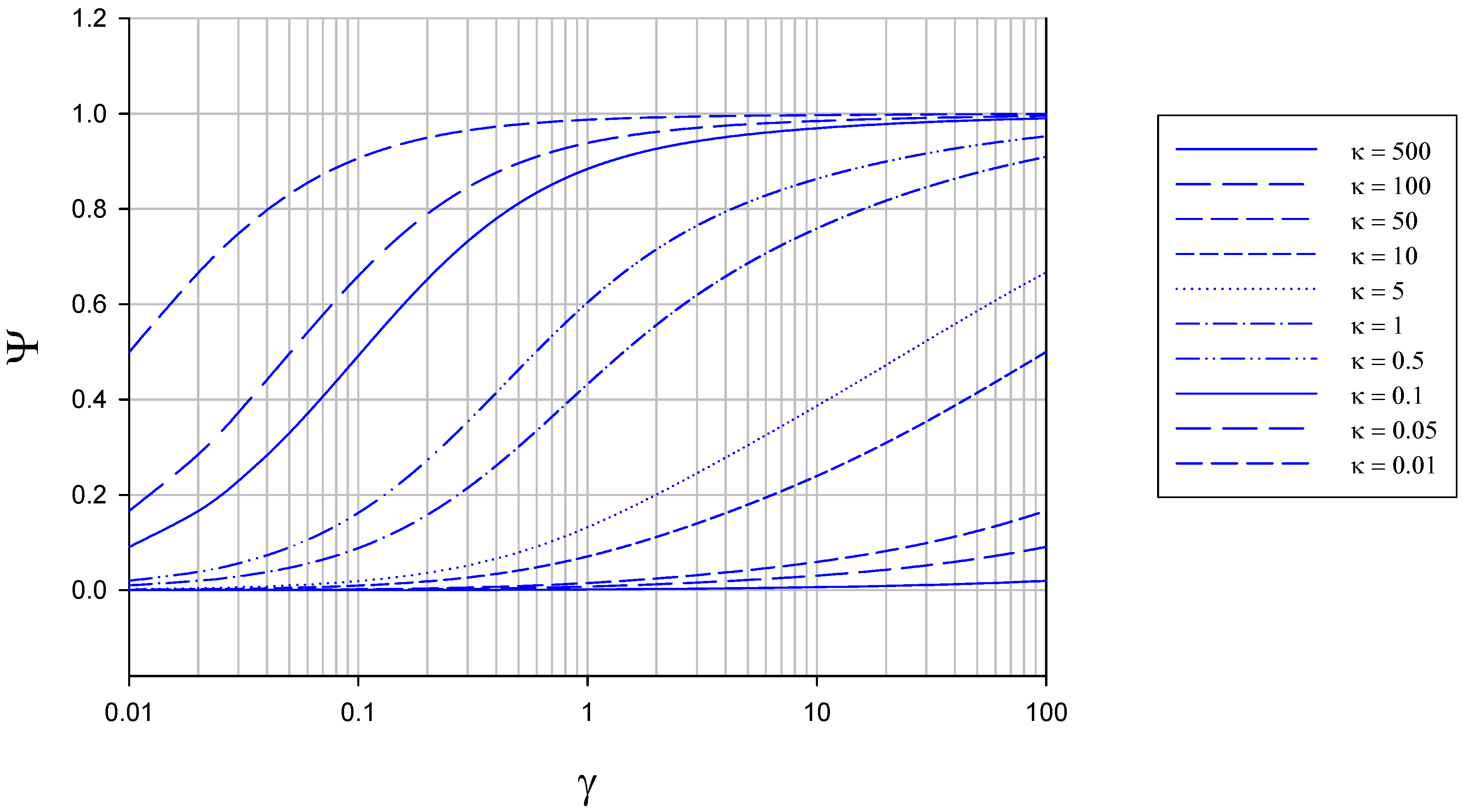{kind=link}
{kind=link}
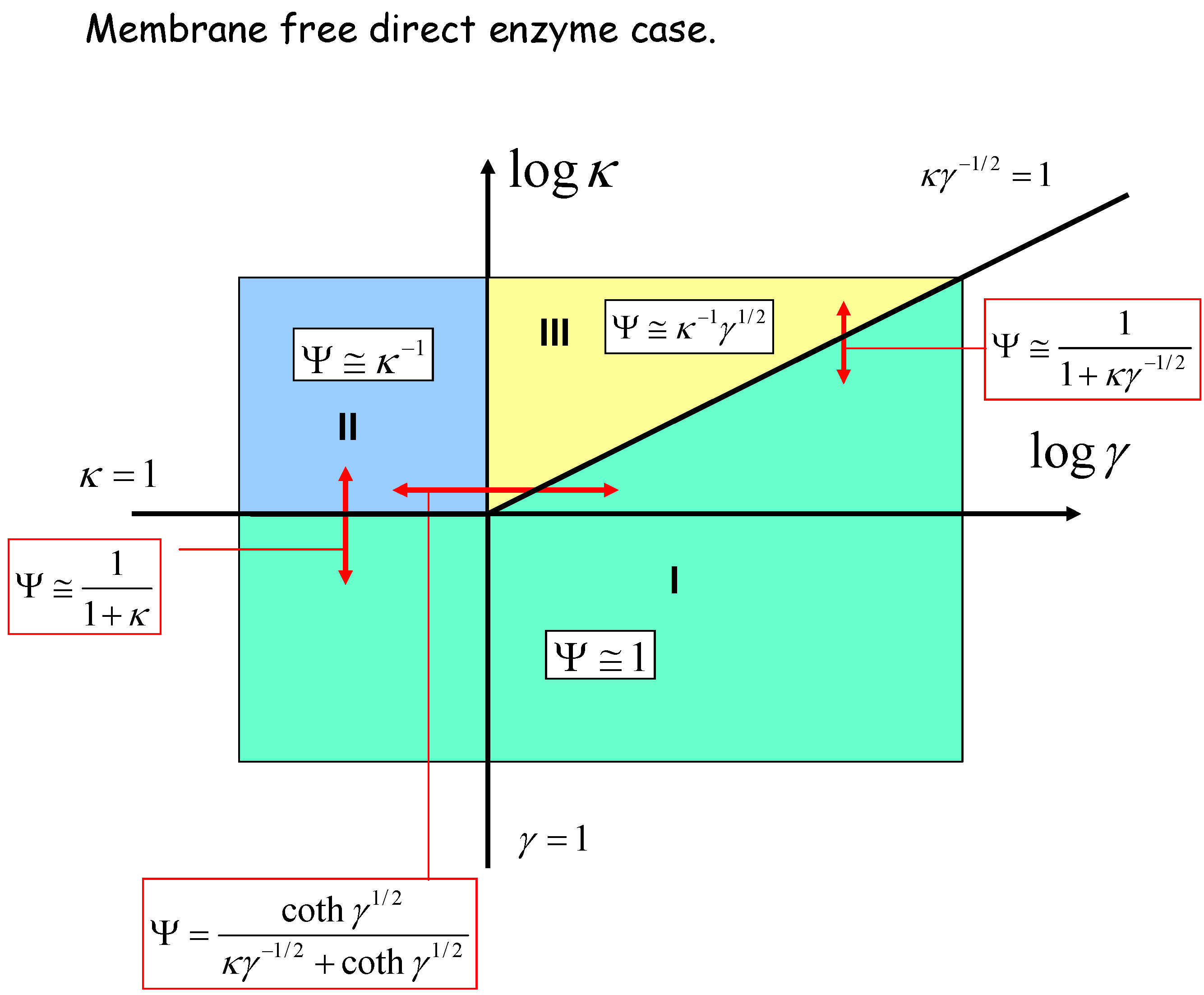{kind=link}
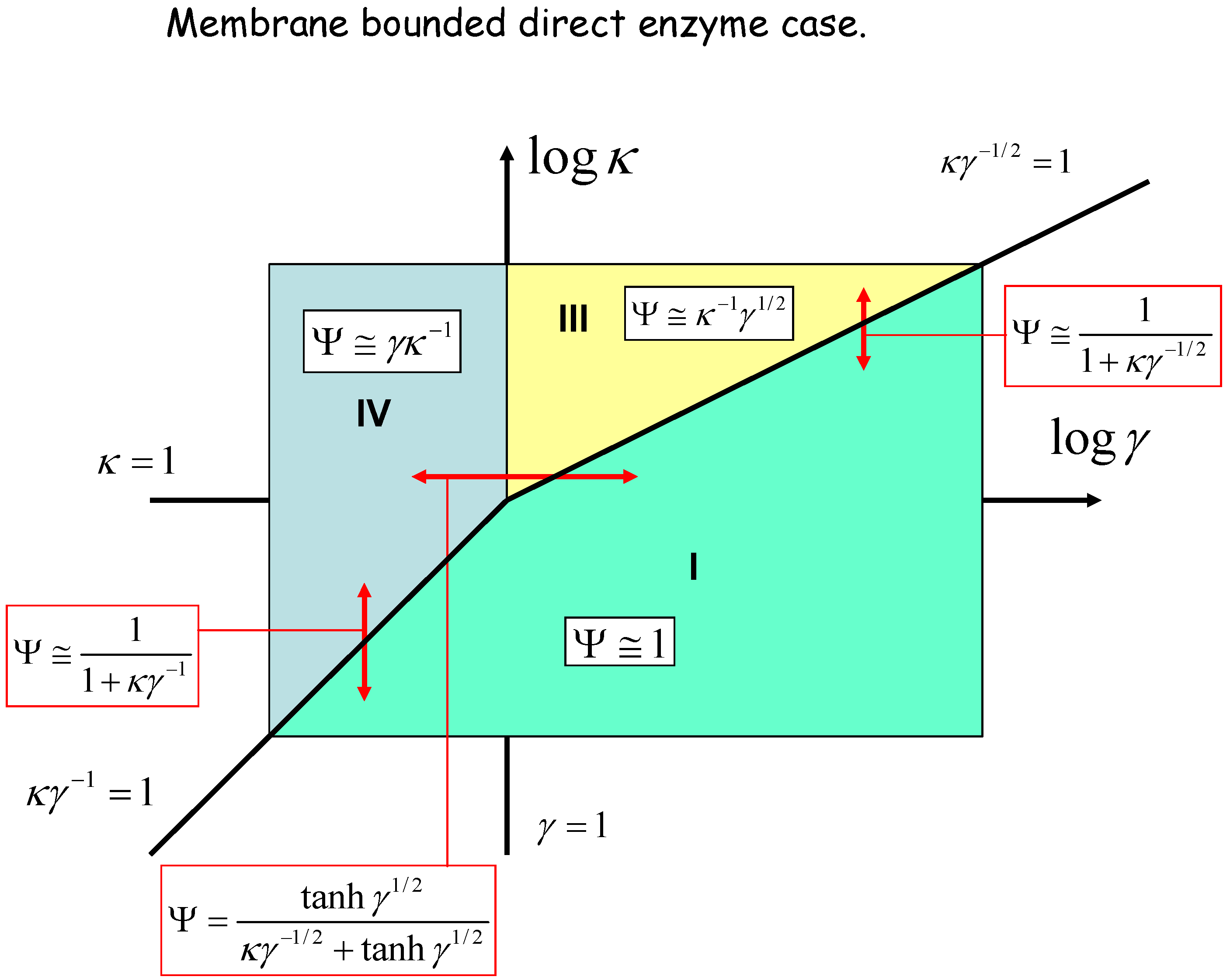{kind=link}
{kind=link}
{kind=link}
| Enzyme modfier | ||||
| Glucose Oxidase | Ferrocene carboxylic acid | Ferrocene Acetic acid | Ferrocene Butanoic acid | |
| E1/2 V(vs SCE) | - ca. 0.41 | 0.3- 0.33 | 0.13-0.18 | 0.09-0.11 |
| # ferrocene per enzyme | 2 | 13 | 22 | 29 |
| kC/s-1 | 800 | 5 | 1100 | 50 |
| KM/mM | 20 | 1 | 5 | 2 |
| kU = kC/KM dm3mol-1s-1 | 40 × 103 | 5 × 103 | 220 × 103 | 25 × 103 |






General comments regarding amperometric enzyme biosensor modelling

Acknowledgements
Glossary of symbols used
| A | Geometric surface area of electrode (units: cm2). |
| a | Concentration of reduced enzyme (units: mol cm-3). |
Normalised parametet quantifying the degree of unsaturation of enzyme/substrate reaction kinetics. | |
| b | Concentration of oxidized enzyme (units: mol cm-3). |
Surface concentration of oxidized enzyme (units: mol cm-3). | |
| c | Total mediator concentration (unit: mol cm-3). |
Total mediator concentration in thin film (unit: mol cm-3). | |
Total bulk mediator concentration in solution (unit: mol cm-3). | |
| D | Enzyme diffusion coefficient (units: cm2 s-1). |
| DM | Diffusion coefficient of redox mediator (unit: cm2 s-1). |
| DS | Diffusion coefficient of substrate (unit: cm2 s-1). |
| δ | Nernst diffusion layer thickness (units: cm). |
| Eox, Ered | Oxidised and reduced forms of the redox enzyme. |
Electrode potential and standard electrode potential (units: V). | |
Total enzyme concentration (units: mol cm-3). | |
| F | Faraday Constant (96,500 C mol-1). |
Net reaction flux (rate) for direct enzyme reaction (units: mol cm-2 s-1). | |
Net reaction flux related to the steady state current flow for immobilized enzyme electrode (unit: mol cm-2 s-1). | |
Reaction flux arising from the mediator/substrate reaction (unit: mol cm-2 s-1). | |
Dimensionless parameter which compares the transit time for crossing the diffusion layer with the homogeneous rate constant describing the facility of the enzyme/substrate reaction kinetics. | |
Dimensionless parameter which compares the flux of the mediator/enzyme. reaction with that for mediator diffusion across the immobilizing film. | |
Reaction layer thickness measuring the distance the oxidized enzyme can travel before it reacts with substrate. | |
| L | Total film thickness (unit: cm). |
| n | Number of electrons transferred in reaction. |
Mixed reaction/diffusion parameter. | |
| S | Substrate or reactant species. |
| s | Substrate (reactant) concentration (units: mol cm-3). |
Bulk substrate concentration (units: mol cm-3). | |
Michaelis constant (units: mol cm-3). | |
Pseudo first order rate constant (units: s-1) for Michaelis-Menten enzyme kinetics. | |
Catalytic rate constant in Michaelis-Menten mechanism (units: s-1). | |
Diffusive rate constant for enzyme transport. | |
Heterogeneous electron transfer rate constant for direct reaction of enzyme at electrode surface (unit: cm s-1). | |
Rate constant for reaction between covalently tethered redox relay molecule and reduced enzyme active site (unit: cm s-1). | |
Rate constant for heterogeneous reaction between redox relay and detector electrode (unit: cm s-1). | |
Composite rate constant quantifying reaction between substrate and the catalytically active oxidized form of the redox enzyme. | |
Unsaturated rate constant describing bimolecular kinetics between enzyme and substrate (units: cm3 mol-1 s-1). | |
Equilibrium constant of step i related to internal processes in Michaelis-Menten adduct formation mechanism illustrated in figure 2 of the text. | |
Dimensionless parameter comparing the rate of reduced enzyme reaction at the electrode surface with that of reduced enzyme diffusion to the electrode surface. | |
Partition coefficient of substrate and mediator respectively. | |
Dimensionless substrate concentration. | |
| P | Product species. |
Dimensionless parameter which defines the balance between the mediator/enzyme kinetic flux and the substrate/enzyme kinetic flux. | |
Dimensionless concentration of reduced enzyme | |
Dimensionless concentration of reduced enzyme at electrode surface. | |
Dimensionless concentration of oxidized mediator. | |
Dimensionless concentration of oxidized mediator at electrode surface. | |
| x | Distance variable (unit: cm). |
Non dimensional distance variable. | |
Normalised electrode potential. | |
Normalised reaction flux for enzyme electrode. | |
Normalised flux arising from turnover of mediator by the substrate. | |
Normalised flux at electrode/solution interface. |
References and Notes
- Rosi, N.L.; Mirkin, C.A. Nanostructures in biodiagnostics. Chem. Rev. 2005, 105, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.; Willner, B.; Katz, E. Functional biosensor systems via surface nanoengineering of electronic elements. Reviews in Molecular Biotechnology 2002, 82, 325–335. [Google Scholar] [CrossRef]
- Willner, I.; Katz, E. Integration of layered redox proteins and conductive supports for bioelectronic applications. Angew Chem. Int. Ed. 2000, 39, 1180–1218. [Google Scholar] [CrossRef]
- Willner, I.; Willner, B. Biomaterials integrated with electronic elements: en route to bioelectronics. Trends in Biotechnology 2001, 19, 222–230. [Google Scholar] [CrossRef]
- Davis, J.J.; Morgan, D.A.; Wrathmell, C.L.; Axford, D.N.; Zhao, J.; Wang, N. Molecular bioelectronics. J. Mater. Chem. 2005, 15, 2160–2174. [Google Scholar] [CrossRef]
- Scheller, F.W.; Wollenberger, U.; Lei, C.; Jin, W.; Ge, B.; Lehmann, C.; Lisdat, F.; Fridman, V. Bioelectrocatalysis by redox enzymes at modified electrodes. Reviews in Molecular Biotechnology 2002, 82, 411–424. [Google Scholar] [CrossRef]
- Schuhmann, W. Amperometric enzyme biosensors based on optimized electron transfer pathways and non-manual immobilization procedures. Reviews in Molecular Biotechnology 2002, 82, 425–441. [Google Scholar] [CrossRef]
- Habermuller, K.; Mosbach, M.; Schuhmann, W. Electron transfer mechanisms in amperometric biosensors. Fresenius J. Anal. Chem. 2000, 366, 560–568. [Google Scholar] [PubMed]
- Schmidt, H.L.; Schuhmann, W. Reagentless oxidoreductase sensors. Biosensors & Bioelectronics 1996, 11, 127–135. [Google Scholar]
- Freire, R.S.; Pessoa, C.A.; Mello, L.D.; Kubota, L.T. Direct electron transfer: an approach for electrochemical biosensors with higher selectivity and sensitivity. J. Braz. Chem. Soc. 2003, 14, 230–243. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Tebbutt, P.; Whitaker, R.G. Kinetic aspects of the use of modified electrodes and mediators in bioelectrochemistry. Prog. Reaction Kinetics 1991, 16, 55–155. [Google Scholar]
- Schuhmann, W.; Ohara, T.J.; Schmidt, H.L.; Heller, A. Electron transfer between glucose oxidase and electrodes via redox mediators bound with flexible chains to the enzyme surface. J. Am. Chem. Soc. 1991, 113, 1394–1397. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Whitaker, R.G.; Green, M.J.; Frew, J. Covalent binding of electron relays to glucose oxidase. J. Chem. Soc., Chem. Commun. 1987, 1603–1604. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Pratt, K.F.E. Modelling of processes in enzyme electrodes. Biosensors & Bioelectronics 1993, 8, 451–462. [Google Scholar]
- Lyons, M.E.G. Electrocatalysis using electroactive polymer films. In Electroactive Polymer Electrochemistry. Part 1. Fundamentals; Lyons, M.E.G., Ed.; Plenum Press: New York, 1994; Chapter 2; pp. 237–374. [Google Scholar]
- Lyons, M.E.G.; Lyons, C.H.; Michas, A.; Bartlett, P.N. Heterogeneous redox catalysis at hydrated oxide layers. J. Electroanal. Chem. 1993, 351, 245–258. [Google Scholar] [CrossRef]
- Albery, W.J.; Knowles, J.R. Evolution of enzyme function and development of catalytic efficiency. Biochemistry 1976, 15, 5631–5640. [Google Scholar] [CrossRef] [PubMed]
- Northrop, D.B. On the meaning of KM and v/KM in enzyme kinetics. J. Chem. Ed. 1998, 75, 1153–1157. [Google Scholar] [CrossRef]
- Albery, W.J.; Bartlett, P.N.; Craston, D.H. Amperometric enzyme electrodes. Part 2. Conducting salts as electrode materials for the oxidation of glucose oxidase. J. Electroanal. Chem. 1985, 194, 223–235. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Cooper, J.M. A review of the immobilization of enzymes in electropolymerized films. J. Electroanal. Chem. 1993, 362, 1–12. [Google Scholar] [CrossRef]
- Gerard, M.; Chaubrey, A.; Malhotra, B.D. Applications of conducting polymers to biosensors. Biosensors & Bioelectronics 2002, 17, 345–359. [Google Scholar]
- Chaubrey, A.; Malhotra, B.D. Mediated biosensors. Biosensors & Bioelectronics. 2002, 17, 441–456. [Google Scholar]
- Mell, L.D.; Maloy, J.T. A model for the amperometric enzyme electrode obtained through digital simulation and applied to the immobilized glucose oxidase system. Anal. Chem. 1975, 47, 299–307. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Whitaker, R.G. Electrochemical immobilization of enzymes. Part 1. Theory. J. Electroanal. Chem. 1987, 224, 27–35. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Whitaker, R.G. Electrochemical immobilization of enzymes. Part 2. Glucose oxidase immobilized in poly-N-methyl pyrrole. J. Electroanal. Chem. 1987, 224, 37–48. [Google Scholar] [CrossRef]
- Gooding, J.J.; Hall, E.A.H.; Hibbert, D.B. From thick films to monolayer recognition layers in amperometric enzyme electrodes. Electroanalysis 1998, 10, 1130–1136. [Google Scholar] [CrossRef]
- Marchesiello, M.; Genies, E. A theoretical model for an amperometric glucose sensor using polypyrroleas an immobilization matrix. J. Electroanal. Chem. 1993, 358, 35–48. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Pratt, K.F.E. Theoretical treatment of diffusion and kinetics in amperometric immobilized enzyme electrodes. Part 1. Redox mediator entrapped within the film. J. Electroanal. Chem. 1995, 397, 61–78. [Google Scholar] [CrossRef]
- Karube, I.; Vokoyama, K.; Tamiya, E. Kinetics of an amperometric glucose sensor with soluble mediator. J. Electroanal. Chem. 1989, 273, 107–117. [Google Scholar]
- Matsumoto, R.; Kano, K.; Ikeda, T. Theory of steady state catalytic currents of mediated bioelectrocatalysis. J. Electroanal. Chem. 2002, 535, 37–40. [Google Scholar] [CrossRef]
- Albery, W.J.; Bartlett, P.N.; Driscoll, B.J.; Lennox, R.B. Amperometric enzyme electrodes. Part 5. The homogeneous mediated mechanism. J. Electroanal. Chem. 1992, 323, 77–102. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Pratt, K.F.E. A study of the kinetics of the reaction between ferrocene monocarboxylic acid and glucose oxidase using the rotating disc electrode. J. Electroanal. Chem. 1995, 397, 53–60. [Google Scholar] [CrossRef]
- Martens, N.; Hall, E.A.H. Model for an immobilized oxidase enzyme electrode in the presence of two oxidants. Anal. Chem. 1994, 66, 2763–2770. [Google Scholar] [CrossRef]
- Coche-Guerente, L.; Desprez, V.; Diard, J.P.; Labbe, P. Amplification of amperometric biosensor responses by electrochemical substrate recycling. Part 1. Theoretical treatment of the catechol-polyphenol oxidase system. J. Electroanal. Chem. 1999, 470, 53–60. [Google Scholar] [CrossRef]
- Baronas, R.; Kulys, J. Ivanauskas, F. Modeling amperometric enzyme electrodes with substrate cyclic conversion. Biosensors & Bioelectronics 2004, 19, 915–922. [Google Scholar]
- Sorochinskii, V.V.; Kurganov, B.I. Steady state kinetics of cyclic conversions of substrate in amperometric bienzyme sensors. Biosensors & Bioelectronics 1996, 11, 225–238. [Google Scholar]
- Limoges, B.; Marchal, D.; Mavre, F.; Saveant, J.M. High amplification rates from the association of two enzymes confined within a nanometric layer immobilized on an electrode: modeling and illustrating example. J. Am. Chem. Soc. 2006, 128, 6014–6015. [Google Scholar] [CrossRef] [PubMed]
- Somasundrum, M.; Tongta, A.; Tanticharoen, M.; Kirtikara, K. A kinetic model for the reduction of enzyme generated hydrogen peroxide at a metal dispersed conducting polymer film. J. Electroanal. Chem. 1997, 440, 259–264. [Google Scholar] [CrossRef]
- Lyons, M.E.G.; McCormack, D.E.; Bartlett, P.N. Microheterogeneous catalysis in modified electrodes. J. Electroanal. Chem. 1989, 261, 51–59. [Google Scholar] [CrossRef]
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Lyons, M.E.G. Modelling the Transport and Kinetics of Electroenzymes at the Electrode/Solution Interface. Sensors 2006, 6, 1765-1790. https://doi.org/10.3390/s6121765
Lyons MEG. Modelling the Transport and Kinetics of Electroenzymes at the Electrode/Solution Interface. Sensors. 2006; 6(12):1765-1790. https://doi.org/10.3390/s6121765
Chicago/Turabian StyleLyons, Michael E.G. 2006. "Modelling the Transport and Kinetics of Electroenzymes at the Electrode/Solution Interface" Sensors 6, no. 12: 1765-1790. https://doi.org/10.3390/s6121765
APA StyleLyons, M. E. G. (2006). Modelling the Transport and Kinetics of Electroenzymes at the Electrode/Solution Interface. Sensors, 6(12), 1765-1790. https://doi.org/10.3390/s6121765