Introduction
Semiconducting tin dioxide is known to exhibit good gas-sensing properties towards various gases [
1]. One of the common ways to tune the sensitivity and selectivity of the materials is to dope them with various noble metals [
2]. A simple, yet promising alternative to enhance the gas-sensing performance is to control the morphological features of the materials during the chemical synthesis. In particular, the generation of high specific surface areas and uniform systems of large (meso)pores will result in a higher probability for a gas to interact with the semiconductor, which is likely to increase the sensitivity of the material.
The concept of utilising self-assembled arrays of amphiphiles to synthesise well-ordered mesoporous materials [
3] has recently been applied to tin(IV) oxides (SnO
2) [
4,
5,
6,
7,
8]. However, only a few of the materials were examined with respect to their gas-sensing properties [6a,8]. Here, we present the synthesis of ordered mesoporous SnO
2 by utilisation of tin(IV) chloride (SnCl
4) as a precursor and cetyltrimethylammonium bromide (CTABr) as a structure-directing agent, as well as the gas sensing properties of the products.
Experimental
In a typical synthesis, a solution of 5.1 g SnCl4·5H2O (98%, Aldrich) in 50 ml water was added to a solution of 6.0 g cetyltrimethylammonium bromide (CTABr, p.a., Merck) in 50 ml water and 4 ml ammonia (25%). The resulting mixture was adjusted to pH 1 by dropwise addition of HCl, stirred for 3 hours, and heated to 100 °C in a Teflon-lined autoclave for five days. The solid product was filtered off, dried and calcined at 300 °C or 450 °C (see below) in order to remove the structure director (CTABr).
X-ray powder diffraction was carried out with a Bruker AXS D8 Advance diffractometer (filtered Cu Kα radiation, 40 kV, 40 mA, 2 seconds counting time per 0.01 °/2θ). Nitrogen physisorption was performed on a Quantachrome Autosorb 6; samples were degassed for 24 hours at 120 °C in vacuum. Transmission electron microscopy (TEM) was carried out with a Philips CM30-ST.
For the preparation of the sensors 140 mg of the mesoporous SnO2 powders were ground and dispersed in 1 ml water. The dispersion was deposited onto substrates (Umweltsensortechnik, UST) with integrated heating and interdigitally structured platinum electrodes, dried at room temperature, and tempered for 60 hours at 350 °C. The gas-sensing properties were measured by means of a gas-mixing equipment using standard mass-flow controllers to provide a well-defined gas flow and a computer to control the experiment and record the resulting data.
Results and discussion
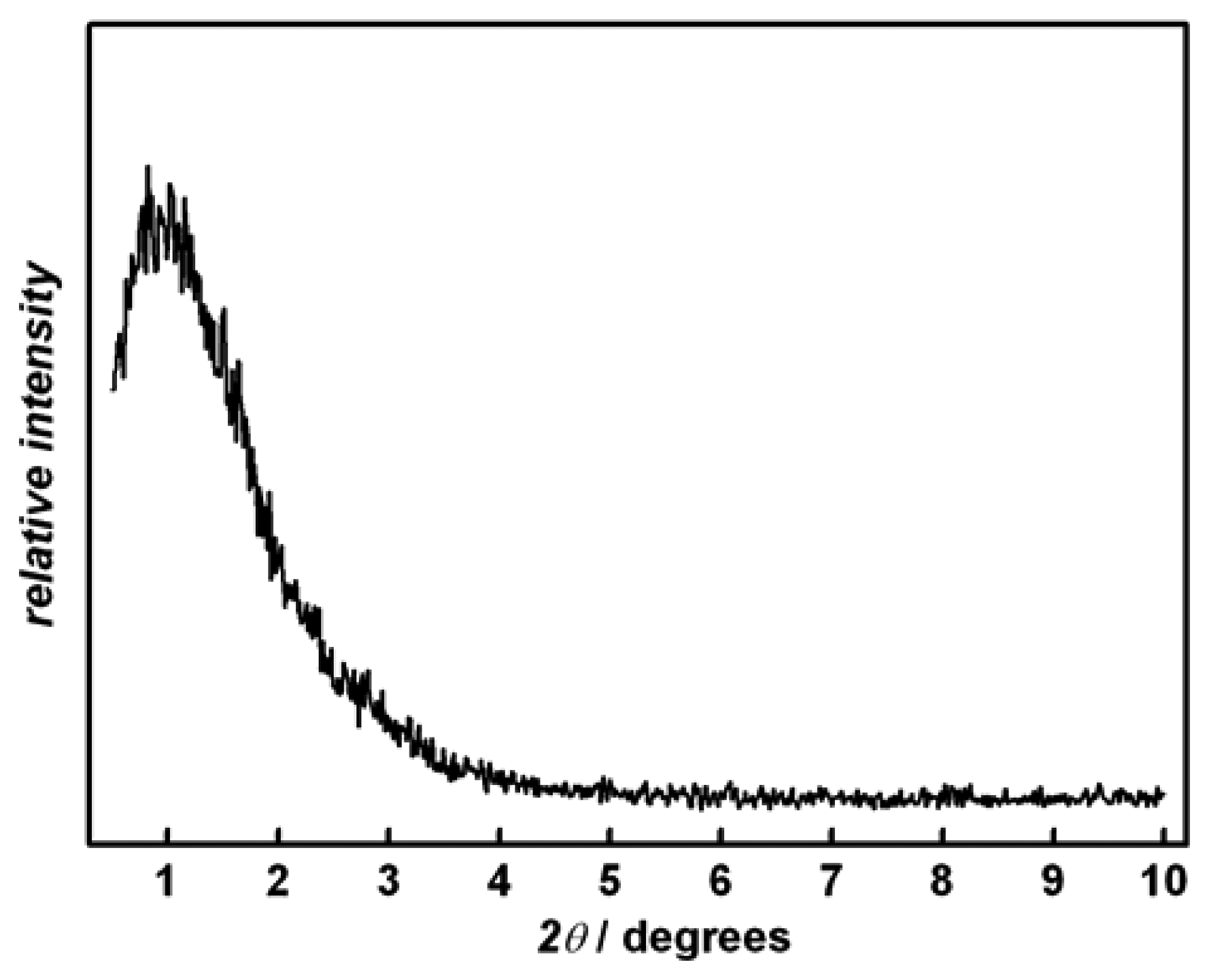
Figure 1 shows the X-ray diffraction (XRD) pattern of a representative sample after calcination at 300 °C. The broad reflection in the low angle region (2
θ = 1 °) is indicative of a disordered mesopore structure; this is supported by TEM (
Figure 2a). The nitrogen physisorption isotherm and the pores size distribution (calculated by the BJH method) of the respective sample is shown in
Figure 3; the specific surface area (BET method) is 160 m
2·g
-1.
A calcination temperature of 300 °C is generally not sufficient for a quantitative removal of CTABr from the pores; accordingly, the samples show a greyish (instead of white) colour after calcination. A higher calcination temperature results in considerable a loss of the mesoscopic structure as evidenced by the disappearance of the XRD reflection and a substantial decrease in the specific surface area (75 m
2·g
-1). However, it was found that all remaining organic residues in the samples calcined at 300 °C are removed upon long-term tempering of the sensors at 350 °C without any loss in mesostructure. This is confirmed by TEM (
Figure 2b) as well as by the absence of bromine as evidenced by EDX.
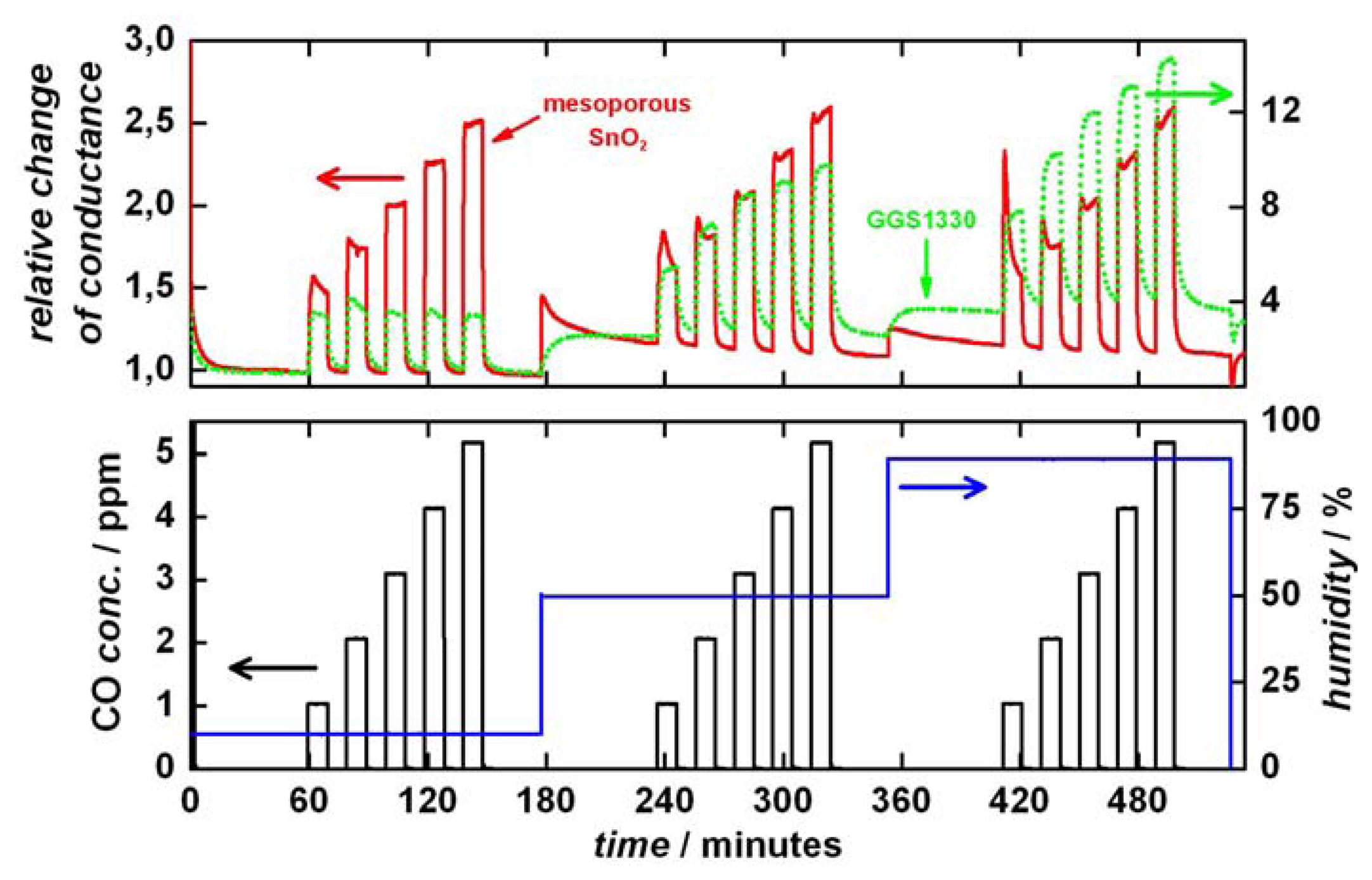
Figure 4 shows the carbon monoxide (CO) gas-sensing performance of a representative mesoporous SnO
2 material as well as of a commercially available SnO
2-based sensor (GGS1330, supplied by UST) for comparison. A constant heating voltage of 4.2 V was applied during the measurements, corresponding to a temperature of about 300 °C. The lower part of the Figure shows the CO concentration as well as the humidity of the test gas. The upper graphs show the relative change in conductance of the two sensors. Note that different scaling is used since the relative change in conductance is up to six times higher for the GGS1330 than for the mesoporous SnO
2 sensors.
The mesoporous SnO
2 sensor shows a very stable base conductance and a quick response to CO. After switching on the CO flow the conductance signal changes from 10 to 90% of the maximum peak height within approximately 10 seconds. After increasing the humidity the base conductance decreases to its original level within 45 minutes. In comparison, the commercial GGS1330 sensor shows a similarly stable base conductance, but a considerably slower response to CO (85 seconds for the 10 to 90% rise). Furthermore, the reaction to the chance in humidity is significantly different, since the conductance does not return to its original base level. Thus, our mesoporous SnO
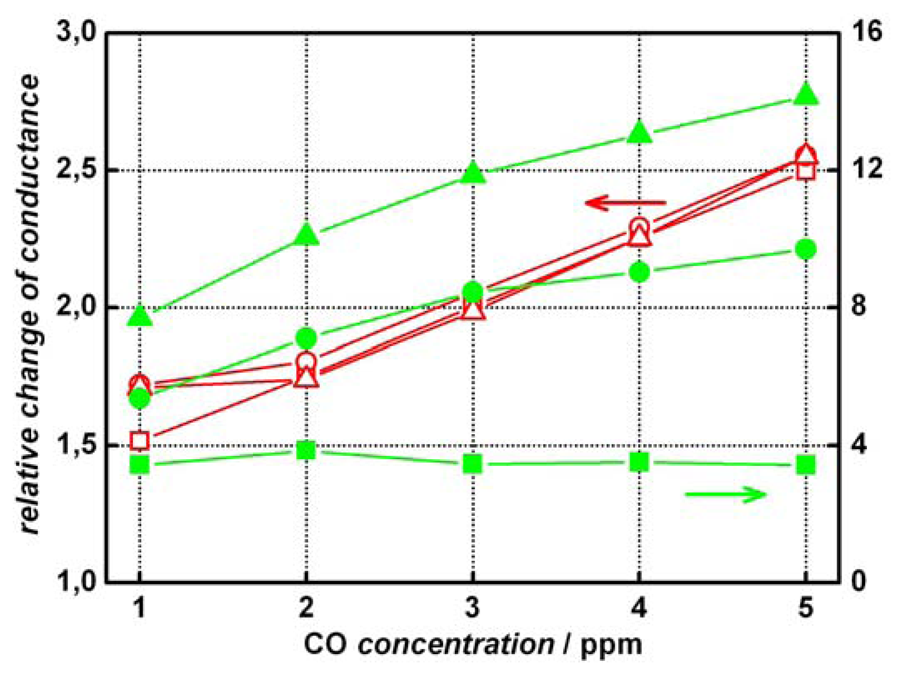
2 sensors show a much higher signal stability against humidity of the test gas. This is shown more clearly in
Figure 5 where the relative change in conductance is plotted against the CO concentration in dependence of the humidity.
Conclusion
In conclusion, we have shown that ordered mesoporous SnO2 powders with large specific surface area can be used for the preparation of gas sensors by application to commercial sensor supports. The materials show a fast and intense response to low concentrations of CO as well as a remarkably strong insensitivity against humidity. Additional measurements on the sensor response to other gases, including studies on the selectivity of our materials, will follow.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




