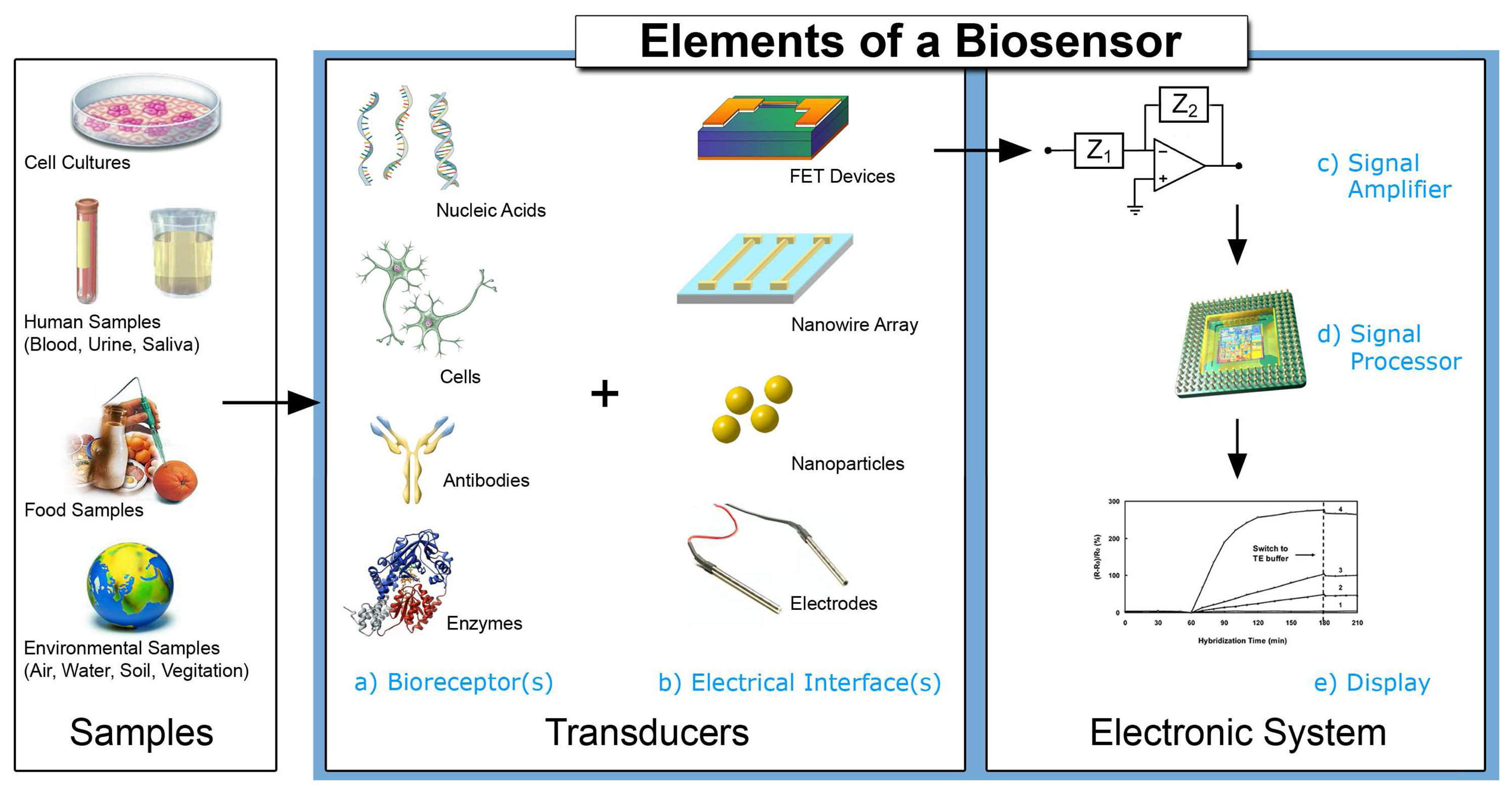
Electrochemical Biosensors - Sensor Principles and Architectures

Abstract
:1. Introduction
- The biocatalyst must be highly specific for the purpose of the analysis, be stable under normal storage conditions and show a low variation between assays.
- The reaction should be as independent as manageable of such physical parameters as stirring, pH and temperature. This will allow analysis of samples with minimal pre-treatment. If the reaction involves cofactors or coenzymes these should, preferably, also be co-immobilized with the enzyme.
- The response should be accurate, precise, reproducible and linear over the concentration range of interest, without dilution or concentration. It should also be free from electrical or other transducer induced noise.
- If the biosensor is to be used for invasive monitoring in clinical situations, the probe must be tiny and biocompatible, having no toxic or antigenic effects. Furthermore, the biosensor should not be prone to inactivation or proteolysis.
- For rapid measurements of analytes from human samples it is desirable that the biosensor can provide real-time analysis.
- The complete biosensor should be cheap, small, portable and capable of being used by semi-skilled operators.
2. Devices
2.1. Electrochemical Detection Techniques
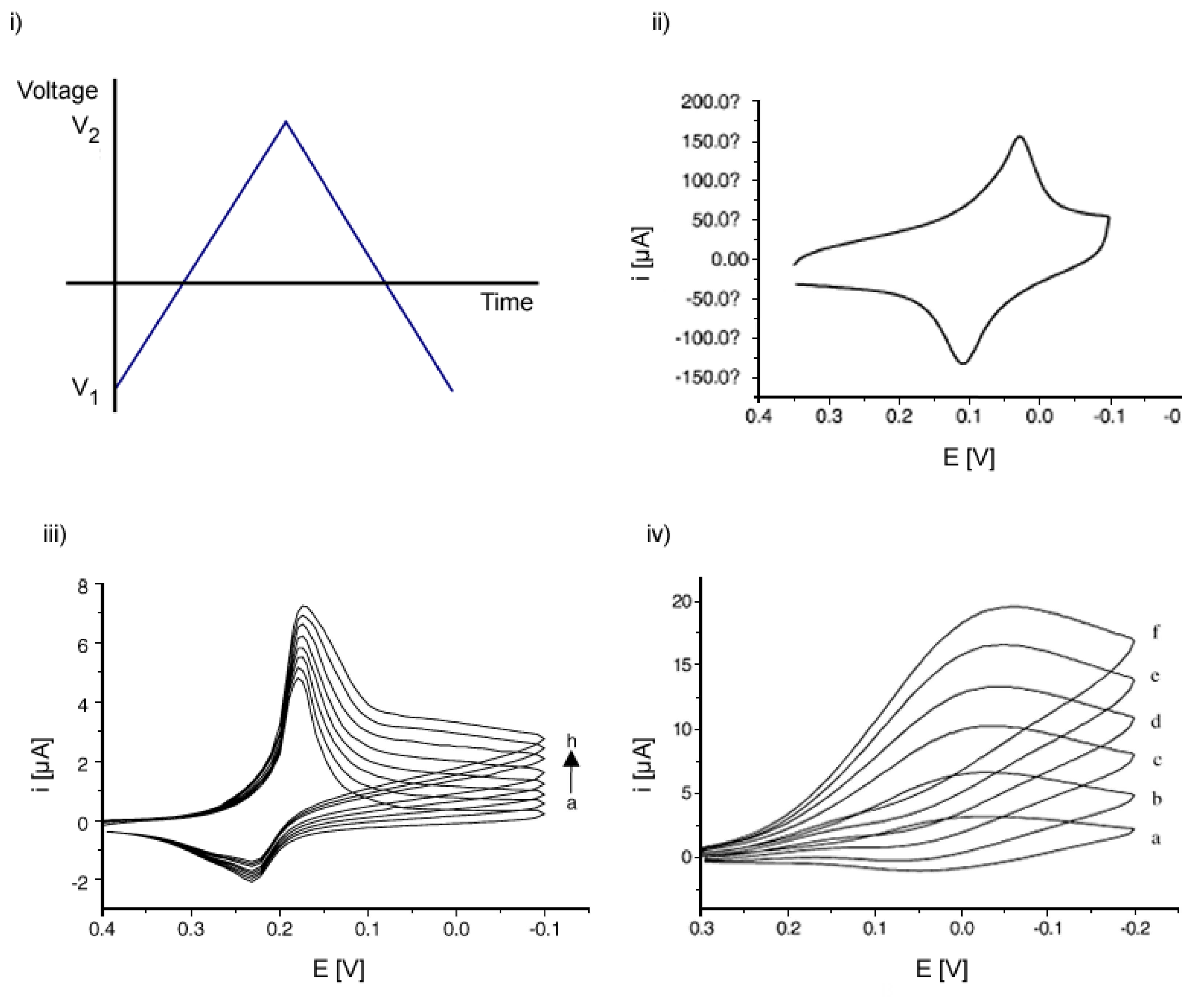
2.1.1. Cyclic Voltammetry (CV)
2.1.2. Chronoamperometry and Chronopotentiometry
2.1.3. Electrochemical Impedance Spectroscopy (EIS)
2.1.4. Field-Effect Transistor (FET)
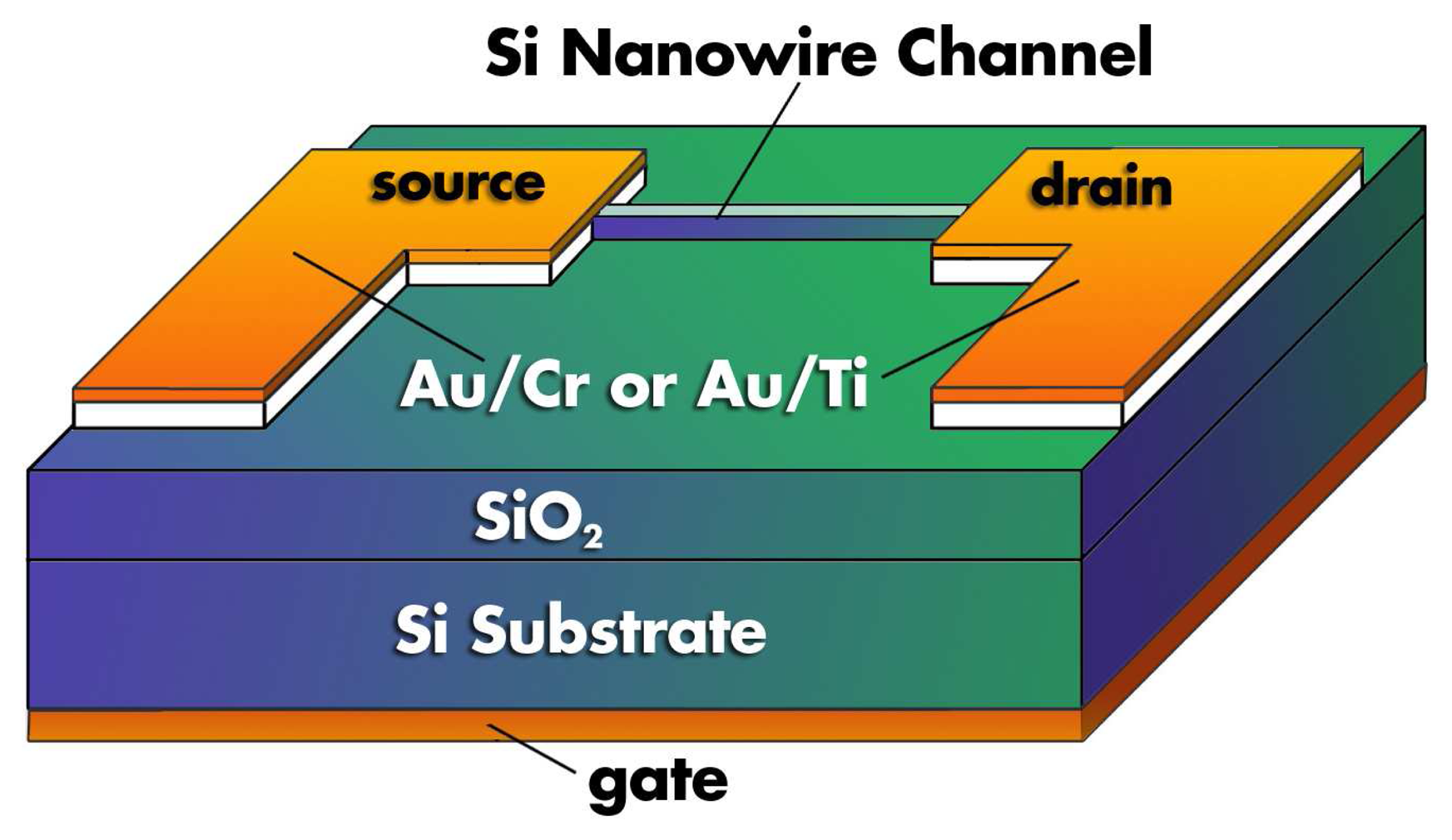
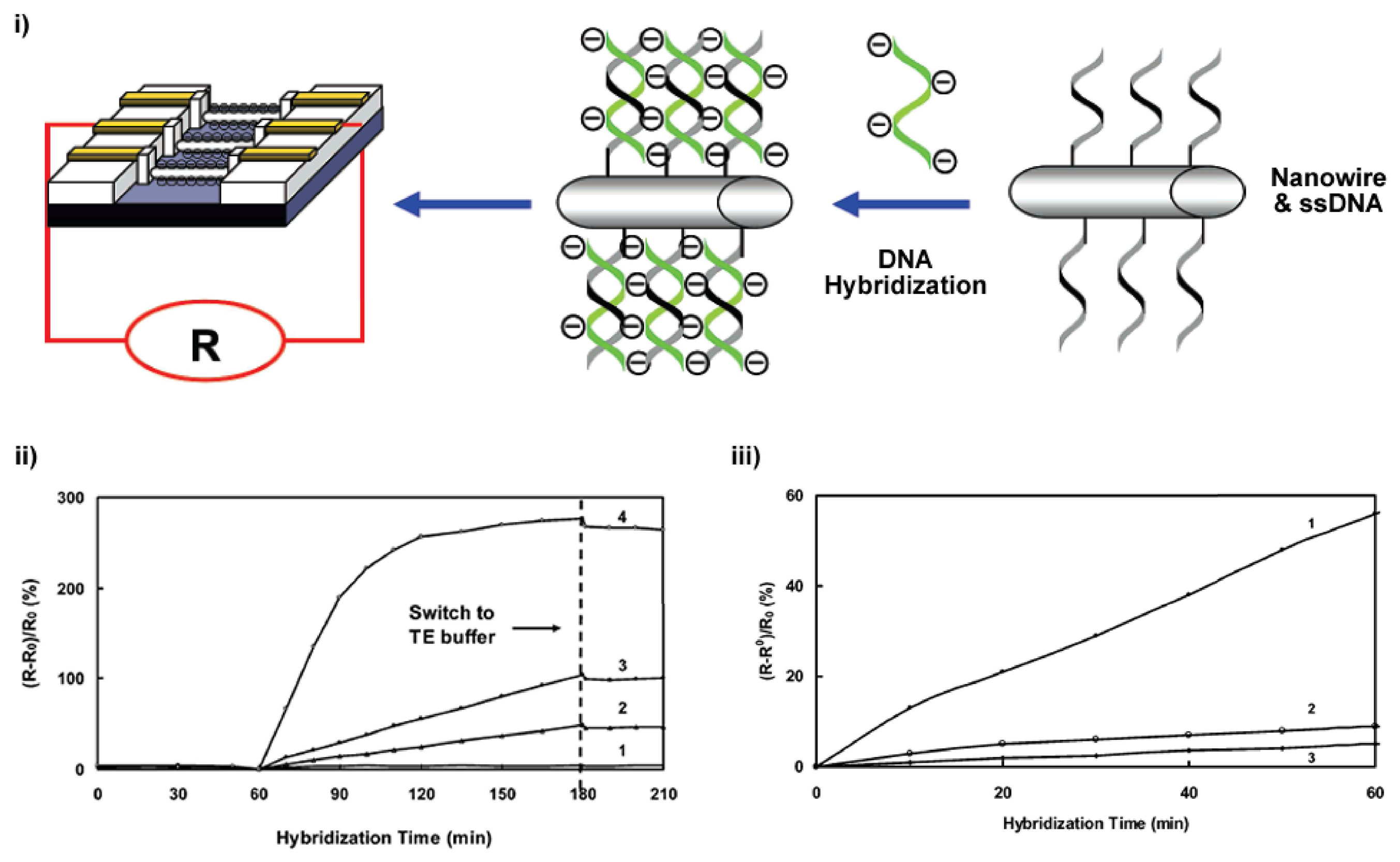
2.2. Nanowires
2.3. Electrochemistry in Combination with Complementary Biosensor Techniques
2.3.1. Electrochemical Surface-Plasmon Resonance (EC-SPR)
2.3.2. Waveguide-Based Techniques and Electrochemistry
2.3.3. Ellipsometry and Electrochemistry
2.3.4. Electrochemical Quartz Crystal Microbalance with Dissipation monitoring (EC-QCM-D)
2.3.5. Scanning Probe Microscopy (SPM)
2.4. Magnetic Biosensors
3. Surface Architecture
3.1. Surface Materials and Modifications
3.2. Electrochemical Signal Transduction
3.3. Enzymes
3.4. Recognition Elements
3.4.1. Antibodies
3.4.2. Antibody Fragments
3.4.3. Aptamers
3.5. Encapsulation of Enzymes
3.5.1. Polyelectrolyte Multilayer (PEM) Capsules
3.5.2. Vesicles
3.5.3. Polymeric Micelles
3.5.4. Hydrogel
3.5.5. Sol-Gel
3.6. Supported Lipid Bilayer Sensor Architectures
4. Conclusions & Outlook
Acknowledgments
References
- Lowe, C.R. Biosensors. Trends in Biotechnology 1984, 2(3), 59–65. [Google Scholar]
- Thevenot, D.R.; Toth, K.; Durst, R. A.; Wilson, G. S. Electrochemical biosensors: recommended definitions and classification. Biosensors & Bioelectronics 2001, 16(1-2), 121–131. [Google Scholar]
- Eggins, B. Chemical sensors and biosensors.; Analytical Techniques in the Sciences. John Wiley & Sons: West Sussex, 2002. [Google Scholar]
- Chaubey, A.; Malhotra, B.D. Mediated biosensors. Biosensors & Bioelectronics 2002, 17(6-7), 441–456. [Google Scholar]
- Kasemo, B. Biological surface science. Surface Science 2002, 500(1-3), 656–677. [Google Scholar]
- Luppa, P.B.; Sokoll, L. J.; Chan, D. W. Immunosensors - principles and applications to clinical chemistry. Clinica Chimica Acta 2001, 314(1-2), 1–26. [Google Scholar]
- Clark, L.C.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Annals of the New York Academy of Sciences 1962, 102(1), 29, –&. [Google Scholar]
- Fang, A.P.; Ng, H. T.; Li, S. F.Y. A high-performance glucose biosensor based on monomolecular layer of glucose oxidase covalently immobilised on indium-tin oxide surface. Biosensors & Bioelectronics 2003, 19(1), 43–49. [Google Scholar]
- Wilson, M.S. Electrochemical immunosensors for the simultaneous detection of two tumor markers. Analytical Chemistry 2005, 77(5), 1496–1502. [Google Scholar]
- D'Orazio, P. Biosensors in clinical chemistry. Clinica Chimica Acta 2003, 334(1-2), 41–69. [Google Scholar]
- Schoning, M.J.; Poghossian, A. Recent advances in biologically sensitive field-effect transistors (biofets). Analyst 2002, 127(9), 1137–1151. [Google Scholar]
- Clark, L.C. Monitor and control of blood and tissue oxygen tensions. Transactions American Society for Artificial Internal Organs 1956, 2, 41, –&. [Google Scholar]
- Clark, L.C.; Clark, E. W. A personalized history of the clark oxygen-electrode. International Anesthesiology Clinics 1987, 25(3), 1–29. [Google Scholar]
- Wang, J. Glucose biosensors: 40 years of advances and challenges. Electroanalysis 2001, 13(12), 983–988. [Google Scholar]
- Mirsky, V.M.; Riepl, M.; Wolfbeis, O. S. Capacitive monitoring of protein immobilization and antigen-antibody reactions on monomolecular alkylthiol films on gold electrodes. Biosensors & Bioelectronics 1997, 12(9-10), 977–989. [Google Scholar]
- Guiseppi-Elie, A.; Lingerfelt, L. Impedimetric detection of dna hybridization: Towards near-patient dna diagnostics. In Immobilisation of DNA on Chips I, volume 260 of Topics in Current Chemistry; pp. 161–186. Springer-Verlag Berlin: Berlin, 2005. [Google Scholar]
- Ben, A.M.; Korpan, Y.; Gonchar, M.; El'skaya, A.; Maaref, M. A.; Jaffrezic-Renault, N.; Martelet, C. Formaldehyde assay by capacitance versus voltage and impedance measurements using bi-layer bio-recognition membrane. Biosensors & Bioelectronics 2006, 22(5), 575–581. [Google Scholar]
- Mehrvar, M.; Abdi, M. Recent developments, characteristics, and potential applications of electrochemical biosensors. Analytical Sciences 2004, 20(8), 1113–1126. [Google Scholar]
- Patolsky, F.; Zheng, G.F.; Lieber, C. M. Nanowire-based biosensors. Analytical Chemistry 2006, 78(13), 4260–4269. [Google Scholar]
- Nair, P.; Alam, M. Dimensionally frustrated diffusion towards fractal adsorbers. Physical Review Letters 2007, 99(25), 256101. [Google Scholar]
- Merkoci, A. Electrochemical biosensing with nanoparticles. Febs Journal 2007, 274(2), 310–316. [Google Scholar]
- Park, S.J.; Taton, T. A.; Mirkin, C. A. Array-based electrical detection of dna with nanoparticle probes. Science 2002, 295(5559), 1503–1506. [Google Scholar]
- Wanekaya, A.K.; Chen, W.; Myung, N. V.; Mulchandani, A. Nanowire-based electrochemical biosensors. Electroanalysis 2006, 18(6), 533–550. [Google Scholar]
- Stadler, B.; Solak, H.H.; Frerker, S.; Bonroy, K.; Frederix, F.; Voros, J.; Grandin, H. M. Nanopatterning of gold colloids for label-free biosensing. Nanotechnology 2007, 18(15), 6. [Google Scholar]
- Wang, J. Electrochemical biosensors: Towards point-of-care cancer diagnostics. Biosensors & Bioelectronics 2006, 21(10), 1887–1892. [Google Scholar]
- Santandreu, M.; Alegret, S.; Fabregas, E. Determination of beta-hcg using amperometric immunosensors based on a conducting immunocomposite. Analytica Chimica Acta 1999, 396(2-3), 181–188. [Google Scholar]
- Yu, D.H.; Blankert, B.; Bodoki, E.; Bollo, S.; Vire, J. C.; Sandulescu, R.; Nomura, A.; Kauffmann, J. M. Amperometric biosensor based on horseradish peroxidase-immobilised magnetic microparticles. Sensors and Actuators B-Chemical 2006, 113(2), 749–754. [Google Scholar]
- Kueng, A.; Kranz, C.; Mizaikoff, B. Amperometric atp biosensor based on polymer entrapped enzymes. Biosensors & Bioelectronics 2004, 19(10), 1301–1307. [Google Scholar]
- Bakker, E.; Pretsch, E. Potentiometric sensors for trace-level analysis. Trac-Trends in Analytical Chemistry 2005, 24(3), 199–207. [Google Scholar]
- Buerk, D. Biosensors. Theory and Applications; Technomic Publishing: Lancaster, 1993. [Google Scholar]
- Caras, S.; Janata, J. Field-effect transistor sensitive to penicillin. Analytical Chemistry 1980, 52(12), 1935–1937. [Google Scholar]
- Hafeman, D.G.; Parce, J. W.; McConnell, H. M. Light-addressable potentiometric sensor for biochemical systems. Science 1988, 240(4856), 1182–1185. [Google Scholar]
- Mourzina, Y.; Yoshinobu, T.; Schubert, J.; Luth, H.; Iwasaki, H.; Schoning, M.J. Ion-selective light-addressable potentiometric sensor (laps) with chalcogenide thin film prepared by pulsed laser deposition. Sensors and Actuators B-Chemical 2001, 80(2), 136–140. [Google Scholar]
- Poghossian, A.; Yoshinobu, T.; Simonis, A.; Ecken, H.; Luth, H.; Schoning, M.J. Penicillin detection by means of field-effect based sensors: Enfet, capacitive eis sensor or laps? Sensors and Actuators B-Chemical 2001, 78(1-3), 237–242. [Google Scholar]
- Xu, G.; Ye, X.; Qin, L.; Xu, Y.; Li, Y.; Li, R.; Wang, P. Cell-based biosensors based on light-addressable potentiometric sensors for single cell monitoring. Biosensors & Bioelectronics 2005, 20, 17571763. [Google Scholar]
- Kloock, J.P.; Moreno, L.; Bratov, A.; Huachupoma, S.; Xu, J.; Wagner, T.; Yoshinobu, T.; Ermolenko, Y.; Vlasov, Y. G.; Schoning, M. J. Pld-prepared cadmium sensors based on chalcogenide glasses- isfet, laps and mu ise semiconductor structures. Sensors and Actuators B-Chemical 2006, 118(1-2), 149–155. [Google Scholar]
- Stein, B.; George, M.; Gaub, H.E.; Parak, W. J. Extracellular measurements of averaged ionic currents with the light-addressable potentiometric sensor (laps). Sensors and Actuators B-Chemical 2004, 98(2-3), 299–304. [Google Scholar]
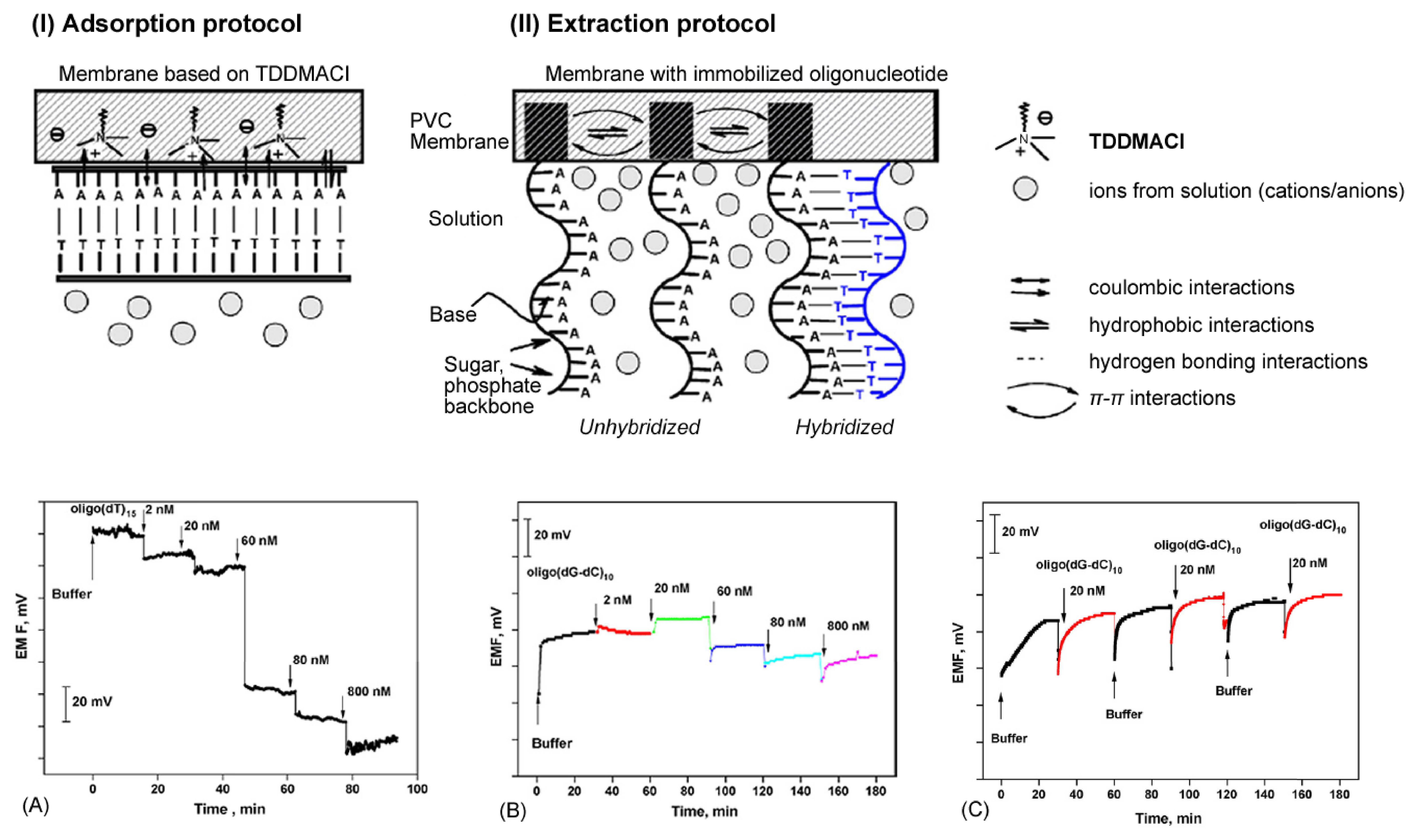
- Shishkanova, T.V.; Volf, R.; Krondak, M.; Kral, V. Functionalization of pvc membrane with ss oligonucleotides for a potentiometric biosensor. Biosensors & Bioelectronics 2007, 22(11), 2712–2717. [Google Scholar]
- Cullen, D.C.; Sethi, R. S.; Lowe, C. R. Multianalyte miniature conductance biosensor. Analytica Chimica Acta 1990, 231(1), 33–40. [Google Scholar]
- Contractor, A.Q.; Sureshkumar, T. N.; Narayanan, R.; Sukeerthi, S.; Lal, R.; Srinivasa, R. S. Conducting polymer-based biosensors. Electrochimica Acta 1994, 39(8-9), 1321–1324. [Google Scholar]
- Yagiuda, K.; Hemmi, A.; Ito, S.; Asano, Y.; Fushinuki, Y.; Chen, C.Y.; Karube, I. Development of a conductivity-based immunosensor for sensitive detection of methamphetamine (stimulant drug) in human urine. Biosensors & Bioelectronics 1996, 11(8), 703–707. [Google Scholar]
- Chouteau, C.; Dzyadevych, S.; Chovelon, J.M.; Durrieu, C. Development of novel conductometric biosensors based on immobilised whole cell chlorella vulgaris microalgae. Biosensors & Bioelectronics 2004, 19(9), 1089–1096. [Google Scholar]
- Heyrovsky, J. The development of polarographic analysis. Analyst 1956, 81(961), 189, –&. [Google Scholar]
- Katz, E.; Willner, I. Probing biomolecular interactions at conductive and semiconductive surfaces by impedance spectroscopy: Routes to impedimetric immunosensors, dna-sensors, and enzyme biosensors. Electroanalysis 2003, 15(11), 913–947. [Google Scholar]
- Pei, R.J.; Cheng, Z. L.; Wang, E. K.; Yang, X. R. Amplification of antigen-antibody interactions based on biotin labeled protein-streptavidin network complex using impedance spectroscopy. Biosensors & Bioelectronics 2001, 16(6), 355–361. [Google Scholar]
- Gosser, D. K. Cyclic Voltammetry, Simulation and Analysis of Reaction Mechanisms.; VHC Publishers, Inc., 1994. [Google Scholar]
- Li, J.P.; Peng, T. Z.; Peng, Y. Q. A cholesterol biosensor based on entrapment of cholesterol oxidase in a silicic sol-gel matrix at a prussian blue modified electrode. Electroanalysis 2003, 15(12), 1031–1037. [Google Scholar]
- Liu, Y.; Yuan, R.; Chai, Y.Q.; Tang, D. P.; Dai, J. Y.; Zhong, X. Direct electrochemistry of horseradish peroxidase immobilized on gold colloid/cysteine/nafion-modified platinum disk electrode. Sensors and Actuators B-Chemical 2006, 115(1), 109–115. [Google Scholar]
- Patolsky, F.; Zayats, M.; Katz, E.; Willner, I. Precipitation of an insoluble product on enzyme monolayer electrodes for biosensor applications: Characterization by faradaic impedance spectroscopy, cyclic voltammetry, and microgravimetric quartz crystal microbalance analyses. Analytical Chemistry 1999, 71(15), 3171–3180. [Google Scholar]
- Nahir, T.M.; Buck, R. P. Modified cottrell behavior in thin-layers - applied voltage steps under diffusion control for constant-resistance systems. Journal of Electroanalytical Chemistry 1992, 341(1-2), 1–14. [Google Scholar]
- Michael, D.J.; Wightman, R. M. Electrochemical monitoring of biogenic amine neurotransmission in real time. Journal of Pharmaceutical and Biomedical Analysis 1999, 19(1-2), 33–46. [Google Scholar]
- Martins, M. C. L.; Fonseca, C.; Barbosa, M. A.; Ratner, B. D. Albumin adsorption on alkanethiols self-assembled monolayers on gold electrodes studied by chronopotentiometry. Biomaterials 2003, 24(21), 3697–3706. [Google Scholar]
- Lorenz, W.; Schulze, K.D. Application of transform-impedance spectrometry. Journal of Electro-analytical Chemistry 1975, 65(1), 141–153. [Google Scholar]
- Willner, I.; Katz, E. e. Bioelectronics.Wiley-VCH, 1st edition; 2005. [Google Scholar]
- Tlili, A.; Abdelghani, A.; Ameur, S.; Jaffrezic-Renault, N. Impedance spectroscopy and affinity measurement of specific antibody-antigen interaction. Materials Science & Engineering C-Biomimetic and Supramolecular Systems 2006, 26(2-3), 546–550. [Google Scholar]
- Fernandez-Sanchez, C.; McNeil, C.J.; Rawson, K. Electrochemical impedance spectroscopy studies of polymer degradation: application to biosensor development. Trac-Trends in Analytical Chemistry 2005, 24(1), 37–48. [Google Scholar]
- Sumner, C.; Sabot, A.; Turner, K.; Krause, S. A transducer based on enzyme-induced degradation of thin polymer films monitored by surface plasmon resonance. Analytical Chemistry 2000, 72(21), 5225–5232. [Google Scholar]
- Tong, Z.Q.; Yuan, R.; Chai, Y. Q.; Xie, Y.; Chen, S. H. A novel and simple biomolecules immobilization method: Electro-deposition zro2 doped with hrp for fabrication of hydrogen peroxide biosensor. Journal of Biotechnology 2007, 128(3), 567–575. [Google Scholar]
- Bakker, E. Electrochemical sensors. Analytical Chemistry 2004, 76(12), 3285–3298. [Google Scholar]
- Hang, T.C.; Guiseppi-Elie, A. Frequency dependent and surface characterization of dna immobilization and hybridization. Biosensors & Bioelectronics 2004, 19(11), 1537–1548. [Google Scholar]
- Patolsky, F.; Katz, E.; Bardea, A.; Willner, I. Enzyme-linked amplified electrochemical sensing of oligonucleotide-dna interactions by means of the precipitation of an insoluble product and using impedance spectroscopy. Langmuir 1999, 15(11), 3703–3706. [Google Scholar]
- Vagin, M.Y.; Karyakin, A. A.; Hianik, T. Surfactant bilayers for the direct electrochemical detection of affinity interactions. Bioelectrochemistry 2002, 56(1-2), 91–93. [Google Scholar]
- Ma, K.S.; Zhou, H.; Zoval, J.; Madou, M. Dna hybridization detection by label free versus impedance amplifying label with impedance spectroscopy. Sensors and Actuators B-Chemical 2006, 114(1), 58–64. [Google Scholar]
- Vlasov, Y.G.; Tarantov, Y. A.; Bobrov, P. V. Analytical characteristics and sensitivity mechanisms of electrolyte-insulator-semiconductor system-based chemical sensors - a critical review. Analytical and Bioanalytical Chemistry 2003, 376(6), 788–796. [Google Scholar]
- Berney, H.; West, J.; Haefele, E.; Alderman, J.; Lane, W.; Collins, J.K. A dna diagnostic biosensor: development, characterisation and performance. Sensors and Actuators B-Chemical 2000, 68(1-3), 100–108. [Google Scholar]
- Horenstein, M. Microelectronic Circuits and Devices; Prentice-Hall: Englewood Cliffs, 1990. [Google Scholar]
- Luo, X.L.; Xu, J. J.; Zhao, W.; Chen, H. Y. Glucose biosensor based on enfet doped with sio2 nanoparticles. Sensors and Actuators B-Chemical 2004, 97(2-3), 249–255. [Google Scholar]
- Schoning, M.J.; Poghossian, A. Bio feds (field-effect devices): State-of-the-art and new directions. Electroanalysis 2006, 18(19-20), 1893–1900. [Google Scholar]
- Errachid, A.; Zine, N.; Samitier, J.; Bausells, J. Fet-based chemical sensor systems fabricated with standard technologies. Electroanalysis 2004, 16(22), 1843–1851. [Google Scholar]
- Poghossian, A.S. Method of fabrication of isfet-based biosensors on an si-sio2-si structure. Sensors and Actuators B-Chemical 1997, 44(1-3), 361–364. [Google Scholar]
- Estrela, P.; Stewart, A.G.; Yan, F.; Migliorato, P. Field effect detection of biomolecular interactions. Electrochimica Acta 2005, 50(25-26), 4995–5000. [Google Scholar]
- Oelssner, W.; Zosel, J.; Guth, U.; Pechstein, T.; Babel, W.; Connery, J.G.; Demuth, C.; Ganseve, M. G.; Verburg, J. B. Encapsulation of isfet sensor chips. Sensors and Actuators B-Chemical 2005, 105(1), 104–117. [Google Scholar]
- Seo, H.I.; Kim, C. S.; Sohn, B. K.; Yeow, T.; Son, M. T.; Haskard, M. Isfet glucose sensor based on a new principle using the electrolysis of hydrogen peroxide. Sensors and Actuators B-Chemical 1997, 40(1), 1–5. [Google Scholar]
- Sohn, B.K.; Cho, B. W.; Kim, C. S.; Kwon, D. H. Isfet glucose and sucrose sensors by using platinum electrode and photo-crosslinkable polymers. Sensors and Actuators B-Chemical 1997, 41(1-3), 7–11. [Google Scholar]
- Poghossian, A.; Luth, H.; Schultze, J.W.; Schoning, M. J. (bio-)chemical and physical microsensor arrays using an identical transducer principle. Electrochimica Acta 2001, 47(1-2), 243–249. [Google Scholar]
- Yeow, T. C. W.; Haskard, M. R.; Mulcahy, D. E.; Seo, H. I.; Kwon, D. H. A very large integrated ph-isfet sensor array chip compatible with standard cmos processes. Sensors and Actuators B-Chemical 1997, 44(1-3), 434–440. [Google Scholar]
- Volotovsky, V.; Kim, N. Cyanide determination by an isfet-based peroxidase biosensor. Biosensors & Bioelectronics 1998, 13(9), 1029–1033. [Google Scholar]
- Kharitonov, A.B.; Zayats, M.; Lichtenstein, A.; Katz, E.; Willner, I. Enzyme monolayer-functionalized field-effect transistors for biosensor applications. Sensors and Actuators B-Chemical 2000, 70(1-3), 222–231. [Google Scholar]
- Zayats, M.; Kharitonov, A.B.; Katz, E.; Buckmann, A. F.; Willner, I. An integrated nad(+)-dependent enzyme-functionalized field-effect transistor (enfet) system: development of a lactate biosensor. Biosensors & Bioelectronics 2000, 15(11-12), 671–680. [Google Scholar]
- Yin, L.T.; Chou, J. C.; Chung, W. Y.; Sun, T. P.; Hsiung, K. P.; Hsiung, S. K. Glucose enfet doped with mno2 powder. Sensors and Actuators B-Chemical 2001, 76(1-3), 187–192. [Google Scholar]
- Park, K.Y.; Choi, S. B.; Lee, M.; Sohn, B. K.; Choi, S. Y. Isfet glucose sensor system with fast recovery characteristics by employing electrolysis. Sensors and Actuators B-Chemical 2002, 83(1-3), 90–97. [Google Scholar]
- Poghossian, A.; Schoning, M.I. Detecting both physical and (bio-)chemical parameters by means of isfet devices. Electroanalysis 2004, 16(22), 1863–1872. [Google Scholar]
- Poghossian, A.; Cherstvy, A.; Ingebrandt, S.; Offenhausser, A.; Schoning, M.J. Possibilities and limitations of label-free detection of dna hybridization with field-effect-based devices. Sensors and Actuators B-Chemical 2005, 111, 470–480. [Google Scholar]
- Koo, S.M.; Edelstein, M. D.; Li, Q. L.; Richter, C. A.; Vogel, E. M. Silicon nanowires as enhancement-mode schottky barrier field-effect transistors. Nanotechnology 2005, 16(9), 1482–1485. [Google Scholar]
- Cui, Y.; Wei, Q.Q.; Park, H. K.; Lieber, C. M. Nanowire nanosensors for highly sensitive and selective detection of biological and chemical species. Science 2001, 293(5533), 1289–1292. [Google Scholar]
- Cui, Y.; Zhong, Z.H.; Wang, D. L.; Wang, W. U.; Lieber, C. M. High performance silicon nanowire field effect transistors. Nano Letters 2003, 3(2), 149–152. [Google Scholar]
- Gao, Z.Q.; Agarwal, A.; Trigg, A. D.; Singh, N.; Fang, C.; Tung, C. H.; Fan, Y.; Buddharaju, K. D.; Kong, J. M. Silicon nanowire arrays for label-free detection of dna. Analytical Chemistry 2007, 79(9), 3291–3297. [Google Scholar]
- Lieber, C.M.; Wang, Z. L. Functional nanowires. Mrs Bulletin 2007, 32(2), 99–108. [Google Scholar]
- Tansil, N.C.; Gao, Z. Q. Nanoparticles in biomolecular detection. Nano Today 2006, 1(1), 28–37. [Google Scholar]
- Xiao, Y.; Patolsky, F.; Katz, E.; Hainfeld, J.F.; Willner, I. Plugging into enzymes: Nanowiring of redox enzymes by a gold nanoparticle. Science 2003, 299(5614), 1877–1881. [Google Scholar]
- Wang, D.H.; Kou, R.; Gil, M. P.; Jakobson, H. P.; Tang, J.; Yu, D. H.; Lu, Y. F. Templated synthesis, characterization, and sensing application of macroscopic platinum nanowire network electrodes. Journal of Nanoscience and Nanotechnology 2005, 5(11), 1904–1909. [Google Scholar]
- Tilke, A.T.; Simmel, F. C.; Lorenz, H.; Blick, R. H.; Kotthaus, J. P. Quantum interference in a one-dimensional silicon nanowire. Physical Review B 2003, 68(7), 6. [Google Scholar]
- Khanal, D. R.; Yim, J. W. L.; Walukiewicz, W.; Wu, J. Effects of quantum confinement on the doping limit of semiconductor nanowires. Nano Letters 2007, 0(0). [Google Scholar]
- Elfstrom, N.; Juhasz, R.; Sychugov, I.; Engfeldt, T.; Karlstrom, A. E.; Linnros, J. Surface charge sensitivity of silicon nanowires: Size dependence. Nano Letters 2007. [Google Scholar]
- Foley, E.L.; Candela, D.; Martini, K. M.; Tuominen, M. T. An undergraduate laboratory experiment on quantized conductance in nanocontacts. American Journal of Physics 1999, 67(5), 389–393. [Google Scholar]
- Landauer, R. Conductance determined by transmission - probes and quantized constriction resistance. Journal of Physics-Condensed Matter 1989, 1(43), 8099–8110. [Google Scholar]
- Agarwal, A.; Sen, D. Conductance of quantum wires: A numerical study of effects of an impurity and interactions. Physical Review B 2006, 73(4), 14. [Google Scholar]
- Das, M.P.; Green, F. Landauer formula without landauer's assumptions. Journal of Physics-Condensed Matter 2003, 15(45), L687–L693. [Google Scholar]
- Patolsky, F.; Timko, B.P.; Zheng, G. F.; Lieber, C. M. Nanowire-based nanoelectronic devices in the life sciences. MRS Bulletin 2007, 32(2), 142–149, Figures 1a & 1c. [Google Scholar]
- Zheng, G.F.; Patolsky, F.; Cui, Y.; Wang, W. U.; Lieber, C. M. Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nature Biotechnology 2005, 23(10), 1294–1301. [Google Scholar]
- Yeh, J.I.; Zimmt, M. B.; Zimmerman, A. L. Nanowiring of a redox enzyme by metallized peptides. Biosensors & Bioelectronics 2005, 21(6), 973–978. [Google Scholar]
- Arwin, H.; Poksinski, M.; Johansen, K. Total internal reflection ellipsometry: principles and applications. Applied Optics 2004, 43(15), 3028–3036. [Google Scholar]
- Elwing, H. Protein absorption and ellipsometry in biomaterial research. Biomaterials 1998, 19(4-5), 397–406. [Google Scholar]
- Goodall, D.G.; Stevens, G. W.; Beaglehole, D.; Gee, M. L. Imaging ellipsometry reflectometry for profiling the shape of a deformable droplet as it approaches an interface. Langmuir 1999, 15(13), 4579–4583. [Google Scholar]
- Pattnaik, P. Surface plasmon resonance - applications in understanding receptor-ligand interaction. Applied Biochemistry and Biotechnology 2005, 126(2), 79–92. [Google Scholar]
- McDonnell, J.M. Surface plasmon resonance: towards an understanding of the mechanisms of biological molecular recognition. Current Opinion in Chemical Biology 2001, 5(5), 572–577. [Google Scholar]
- Liedberg, B.; Nylander, C.; Lundstrom, I. Biosensing with surface-plasmon resonance - how it all started. Biosensors & Bioelectronics 1995, 10(8), R1–R9. [Google Scholar]
- Campagnolo, C.; Meyers, K.J.; Ryan, T.; Atkinson, R. C.; Chen, Y. T.; Scanlan, M. J.; Ritter, G.; Old, L. J.; Batt, C. A. Real-time, label-free monitorine, of tumor antigen and serum antibody interactions. Journal of Biochemical and Biophysical Methods 2004, 61(3), 283–298. [Google Scholar]
- Jung, L.S.; Campbell, C. T.; Chinowsky, T. M.; Mar, M. N.; Yee, S. S. Quantitative interpretation of the response of surface plasmon resonance sensors to adsorbed films. Langmuir 1998, 14(19), 5636–5648. [Google Scholar]
- Badia, A.; Arnold, S.; Scheumann, V.; Zizlsperger, M.; Mack, J.; Jung, G.; Knoll, W. Probing the electrochemical deposition and/or desorption of self-assembled and electropolymerizable organic thin films by surface plasmon spectroscopy and atomic force microscopy. Sensors and Actuators B-Chemical 1999, 54(1-2), 145–165. [Google Scholar]
- De Feijter, J.; Benjamins, J.; Veer, V. Ellipsometry as a tool to study the adsorption behaviour of synthetic and biopolymers at the air-water interface. Biopolymers 1978, 17(7), 1759–1772. [Google Scholar]
- Voros, J.; Ramsden, J.J.; Csucs, G.; Szendro, I.; De Paul, S. M.; Textor, M.; Spencer, N. D. Optical grating coupler biosensors. Biomaterials 2002, 23(17), 3699–3710. [Google Scholar]
- Georgiadis, R.; Peterlinz, K.P.; Peterson, A. W. Quantitative measurements and modeling of kinetics in nucleic acid monolayer films using spr spectroscopy. Journal of the American Chemical Society 2000, 122(13), 3166–3173. [Google Scholar]
- Heaton, R.J.; Peterson, A. W.; Georgiadis, R. M. Electrostatic surface plasmon resonance: Direct electric field-induced hybridization and denaturation in monolayer nucleic acid films and label-free discrimination of base mismatches. Proceedings of the National Academy of Sciences of the United States of America 2001, 98(7), 3701–3704. [Google Scholar]
- Mullett, W.M.; Lai, E. P.C.; Yeung, J. M. Surface plasmon resonance-based immunoassays. Methods 2000, 22(1), 77–91. [Google Scholar]
- Kang, X.F.; Cheng, G. J.; Dong, S. J. A novel electrochemical spr biosensor. Electrochemistry Communications 2001, 3(9), 489–493. [Google Scholar]
- Lavers, C.R.; Harris, R. D.; Hao, S.; Wilkinson, J. S.; Odwyer, K.; Brust, M.; Schiffrin, D. J. Electrochemically-controlled wave-guide-coupled surface-plasmon sensing. Journal of Electroanalytical Chemistry 1995, 387(1-2), 11–22. [Google Scholar]
- Baba, A.; Park, M.K.; Advincula, R. C.; Knoll, W. Simultaneous surface plasmon optical and electrochemical investigation of layer-by-layer self-assembled conducting ultrathin polymer films. Langmuir 2002, 18(12), 4648–4652. [Google Scholar]
- Peterlinz, K.A.; Georgiadis, R. In situ kinetics of self-assembly by surface plasmon resonance spectroscopy. Langmuir 1996, 12(20), 4731–4740. [Google Scholar]
- Georgiadis, R.; Peterlinz, K.A.; Rahn, J. R.; Peterson, A. W.; Grassi, J. H. Surface plasmon resonance spectroscopy as a probe of in-plane polymerization in monolayer organic conducting films. Langmuir 2000, 16(17), 6759–6762. [Google Scholar]
- Peterson, A.W.; Heaton, R. J.; Georgiadis, R. M. The effect of surface probe density on dna hybridization. Nucleic Acids Research 2001, 29(24), 5163–5168. [Google Scholar]
- Wang, F.; Wang, J.L.; Chen, H. J.; Dong, S. J. Assembly process of cuhcf/mpa multilayers on gold nanoparticles modified electrode and characterization by electrochemical spr. Journal of Electroanalytical Chemistry 2007, 600(2), 265–274. [Google Scholar]
- Kurrat, R.; Textor, M.; Ramsden, J.J.; Boni, P.; Spencer, N. D. Instrumental improvements in optical waveguide light mode spectroscopy for the study of biomolecule adsorption. Review of Scientific Instruments 1997, 68(5), 2172–2176. [Google Scholar]
- Bearinger, J.P.; Voros, J.; Hubbell, J. A.; Textor, M. Electrochemical optical waveguide lightmode spectroscopy (ec-owls): A pilot study using evanescent-field optical sensing under voltage control to monitor polycationic polymer adsorption onto indium tin oxide (ito)-coated waveguide chips. Biotechnology and Bioengineering 2003, 82(4), 465–473. [Google Scholar]
- Brusatori, M.A.; Van Tassel, P. R. Biosensing under an applied voltage using optical waveguide lightmode spectroscopy. Biosensors & Bioelectronics 2003, 18(10), 1269–1277. [Google Scholar]
- Ying, P.Q.; Viana, A. S.; Abrantes, L. M.; Jin, G. Adsorption of human serum albumin onto gold: a combined electrochemical and ellipsometric study. Journal of Colloid and Interface Science 2004, 279(1), 95–99. [Google Scholar]
- Wang, Z.H.; Viana, A. S.; Jin, G.; Abrantes, L. M. Immunosensor interface based on physical and chemical immunogylobulin g adsorption onto mixed self-assembled monolayers. Bioelectrochemistry 2006, 69(2), 180–186. [Google Scholar]
- Yu, Y.; Jin, G. Influence of electrostatic interaction on fibrinogen adsorption on gold studied by imaging ellipsometry combined with electrochemical methods. Journal of Colloid and Interface Science 2005, 283(2), 477–481. [Google Scholar]
- Marx, K. Quartz crystal microbalance: A useful tool for studying thin polymer films and complexes biomolecular systems at the solution-surface interface. Bio Macromolecules 2002, 4(5), 1099–1120. [Google Scholar]
- Hook, F.; Rodahl, M.; Brzezinski, P.; Kasemo, B. Measurements using the quartz crystal microbalance technique of ferritin monolayers on methyl-thiolated gold: Dependence of energy dissipation and saturation coverage on salt concentration. Journal of Colloid and Interface Science 1998, 208(1), 63–67. [Google Scholar]
- Hook, F.; Voros, J.; Rodahl, M.; Kurrat, R.; Boni, P.; Ramsden, J.J.; Textor, M.; Spencer, N. D.; Tengvall, P.; Gold, J.; Kasemo, B. A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation. Colloids and Surfaces B-Biointerfaces 2002, 24(2), 155–170. [Google Scholar]
- Dong, Y.G. The frequency response of qcm in electrochemically characterizing the immobilization on gold electrode. Sensors and Actuators B-Chemical 2005, 108(1-2), 622–626. [Google Scholar]
- Schumacher, R. The quartz microbalance - a novel-approach to the insitu investigation of interfacial phenomena at the solid liquid junction. Angewandte Chemie-International Edition in English 1990, 29(4), 329–343. [Google Scholar]
- Beissenhirtz, M.K.; Kafka, B.; Schafer, D.; Wolny, M.; Lisdat, F. Electrochemical quartz crystal microbalance studies on cytochrome c/polyelectrolyte multilayer assemblies on gold electrodes. Electroanalysis 2005, 17(21), 1931–1937. [Google Scholar]
- Bott, A. Characterization of films immobilized on an electrode surface using the electrochemical quartz crystal microbalance. Current Separation 1999, 18(3), 79–83. [Google Scholar]
- Binnig, G.; Rohrer, H. Scanning tunneling microscopy. Helvetica Physica Acta 1982, 55(6), 726–735. [Google Scholar]
- Binnig, G.; Rohrer, H. Scanning tunneling microscopy. Ibm Journal of Research and Development 1986, 30(4), 355–369. [Google Scholar]
- Bae, S.E.; Gewirth, A. A. In situ ec-stm studies of mps, sps, and chloride on cu(100): Structural studies of accelerators for dual damascene electrodeposition. Langmuir 2006, 22(25), 10315–10321. [Google Scholar]
- Colton, R.J.; Baselt, D. R.; Dufrene, Y. F.; Green, J. B.D.; Lee, G. U. Scanning probe microscopy. Current Opinion in Chemical Biology 1997, 1(3), 370–377. [Google Scholar]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Physical Review Letters 1986, 56(9), 930–933. [Google Scholar]
- Rugar, D.; Hansma, P. Atomic force microscopy. Physics Today 1990, 43(10), 23–30. [Google Scholar]
- Jalili, N.; Laxminarayana, K. A review of atomic force microscopy imaging systems: application to molecular metrology and biological sciences. Mechatronics 2004, 14(8), 907–945. [Google Scholar]
- Muguruma, H.; Kase, Y. Structure and biosensor characteristics of complex between glucose oxidase and plasma-polymerized nanothin film. Biosensors & Bioelectronics 2006, 22(5), 737–743. [Google Scholar]
- Tominaga, M.; Ohira, A.; Yamaguchi, Y.; Kunitake, M. Electrochemical, afm and qcm studies on ferritin immobilized onto a self-assembled monolayer-modified gold electrode. Journal of Electroanalytical Chemistry 2004, 566(2), 323–329. [Google Scholar]
- Immoos, C.E.; Lee, S. J.; Grinstaff, M. W. Conformationally gated electrochemical gene detection. Chembiochem 2004, 5(8), 1100–1103. [Google Scholar]
- Dong, Y.Z.; Shannon, C. Heterogeneous immunosensing using antigen and antibody monolayers on gold surfaces with electrochemical and scanning probe detection. Analytical Chemistry 2000, 72(11), 2371–2376. [Google Scholar]
- Bergamini, J.F.; Ghilane, J.; Guilloux-Viry, M.; Hapiot, P. In situ ec-afm imaging of cathodic modifications of platinum surfaces performed in dimethylformamide. Electrochemistry Communications 2004, 6(2), 188–192. [Google Scholar]
- Yamaguchi, Y.; Shiota, M.; Nakayama, Y.; Hirai, N.; Hara, S. Combined in situ ec-afm and cv measurement study on lead electrode for lead-acid batteries. Journal of Power Sources 2001, 93(1-2), 104–111. [Google Scholar]
- Eliaz, N.; Eliyahu, M. Electrochemical processes of nucleation and growth of hydroxyapatite on titanium supported by real-time electrochemical atomic force microscopy. Journal of Biomedical Materials Research Part A 2007, 80A(3), 621–634. [Google Scholar]
- Bard, A.J.; Fan, F. R.F.; Kwak, J.; Lev, O. Scanning electrochemical microscopy - introduction and principles. Analytical Chemistry 1989, 61(2), 132–138. [Google Scholar]
- Kwak, J.; Bard, A.J. Scanning electrochemical microscopy - apparatus and two-dimensional scans of conductive and insulating substrates. Analytical Chemistry 1989, 61(17), 1794–1799. [Google Scholar]
- Kwak, J.; Bard, A.J. Scanning electrochemical microscopy - theory of the feedback mode. Analytical Chemistry 1989, 61(11), 1221–1227. [Google Scholar]
- Sun, P.; Laforge, F.O.; Mirkin, M. V. Scanning electrochemical microscopy in the 21st century. Physical Chemistry Chemical Physics 2007, 9(7), 802–823. [Google Scholar]
- Edwards, M.A.; Martin, S.; Whitworth, A. L.; Macpherson, J. V.; Unwin, P. R. Scanning electrochemical microscopy: principles and applications to biophysical systems. Physiological Measurement 2006, 27(12), R63–R108. [Google Scholar]
- Amemiya, S.; Guo, J.D.; Xiong, H.; Gross, D. A. Biological applications of scanning electrochemical microscopy: chemical imaging of single living cells and beyond. Analytical and Bioanalytical Chemistry 2006, 386(3), 458–471. [Google Scholar]
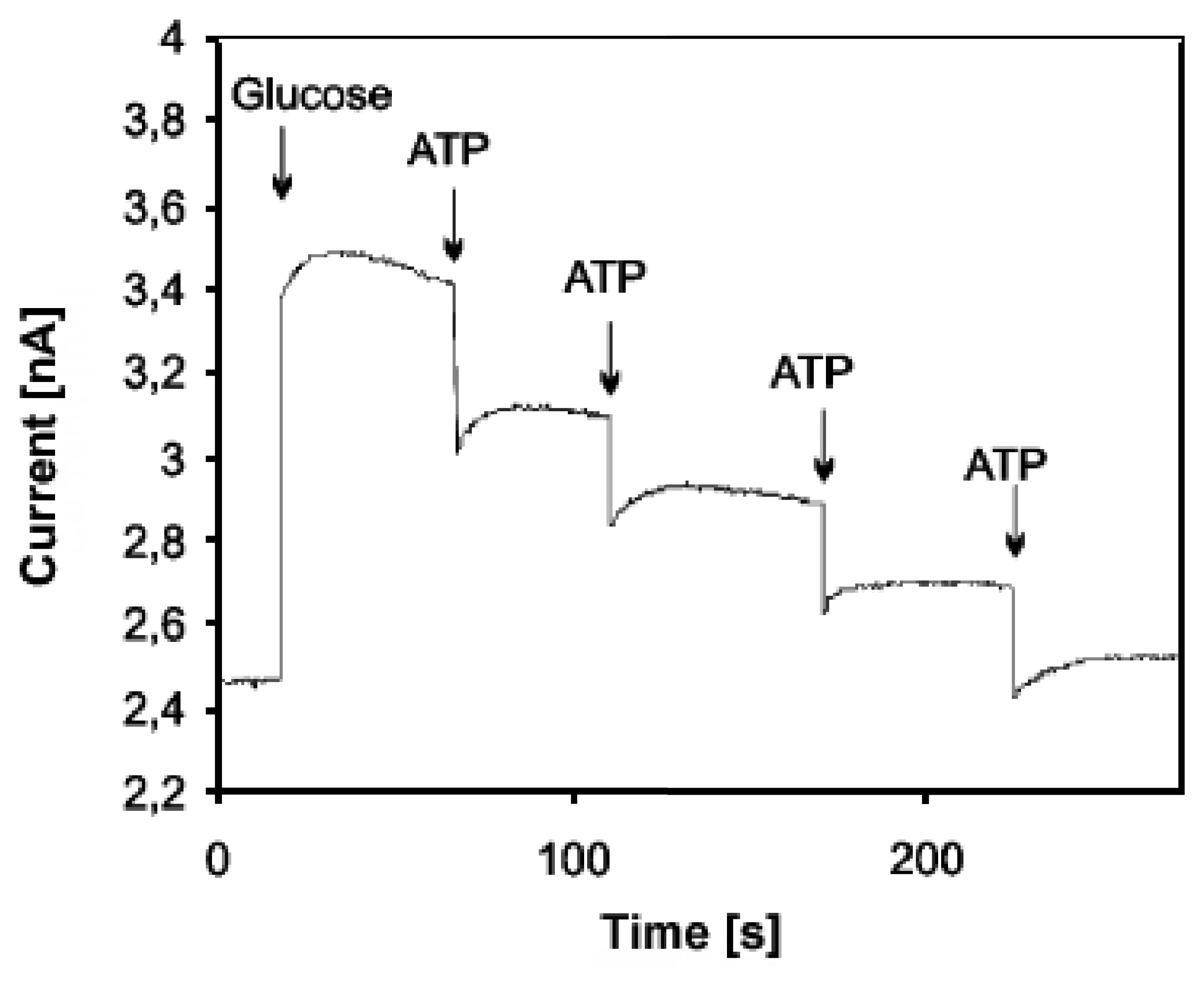
- Kueng, A.; Kranz, C.; Mizaikoff, B. Imaging of atp membrane transport with dual micro-disk electrodes and scanning electrochemical microscopy. Biosensors & Bioelectronics 2005, 21(2), 346–353. [Google Scholar]
- Kueng, A.; Kranz, C.; Lugstein, A.; Bertagnolli, E.; Mizaikoff, B. Afm-tip-integrated amperometric microbiosensors: High-resolution imaging of membrane transport. Angewandte Chemie-International Edition 2005, 44(22), 3419–3422. [Google Scholar]
- Fortin, E.; Defontaine, Y.; Mailley, P.; Livache, T.; Szunerits, S. Micro-imprinting of oligonucleotides and oligonucleotide gradients on gold surfaces: A new approach based on the combination of scanning electrochemical microscopy and surface plasmon resonance imaging (secm/spr-i). Electroanalysis 2005, 17(5-6), 495–503. [Google Scholar]
- Cliffel, D.E.; Bard, A. J. Scanning electrochemical microscopy. 36. a combined scanning electrochemical microscope quartz crystal microbalance instrument for studying thin films. Analytical Chemistry 1998, 70(9), 1993–1998. [Google Scholar]
- Gollas, B.; Bartlett, P.N.; Denuault, G. An instrument for simultaneous eqcm impedance and secm measurements. Analytical Chemistry 2000, 72(2), 349–356. [Google Scholar]
- Xiang, C.H.; Xie, Q. J.; Hu, J. M.; Yao, S. Symmetric current oscillations at tip and substrate electrodes of scanning electrochemical microscope during silver deposition/stripping. Journal of Electroanalytical Chemistry 2005, 584(2), 201–209. [Google Scholar]
- Boldt, F.M.; Heinze, J.; Diez, M.; Petersen, J.; Borsch, M. Real-time ph microscopy down to the molecular level by combined scanning electrochemical microscopy/single-molecule fluorescence spectroscopy. Analytical Chemistry 2004, 76(13), 3473–3481. [Google Scholar]
- Shi, G.D.; Garfias-Mesias, L. F.; Smyrl, W. H. Preparation of a gold-sputtered optical fiber as a microelectrode for electrochemical microscopy. Journal of the Electrochemical Society 1998, 145(6), 2011–2016. [Google Scholar]
- Fan, F.R. F.; Cliffel, D.; Bard, A. J. Scanning electrochemical microscopy. 37. light emission by electrogenerated chemiluminescence at secm tips and their application to scanning optical microscopy. Analytical Chemistry 1998, 70(14), 2941–2948. [Google Scholar]
- Gardner, C.E.; Macpherson, J. V. Atomic force microscopy probes go electrochemical. Analytical Chemistry 2002, 74(21), 576A–584A. [Google Scholar]
- Kurulugama, R.T.; Wipf, D. O.; Takacs, S. A.; Pongmayteegul, S.; Garris, P. A.; Baur, J. E. Scanning electrochemical microscopy of model neurons: Constant distance imaging. Analytical Chemistry 2005, 77(4), 1111–1117. [Google Scholar]
- Macpherson, J.V.; Unwin, P. R. Combined scanning electrochemical-atomic force microscopy. Analytical Chemistry 2000, 72(2), 276–285. [Google Scholar]
- Macpherson, J.V.; Unwin, P. R. Noncontact electrochemical imaging with combined scanning electrochemical atomic force microscopy. Analytical Chemistry 2001, 73(3), 550–557. [Google Scholar]
- Gardner, C.E.; Unwin, P. R.; Macpherson, J. V. Correlation of membrane structure and transport activity using combined scanning electrochemical-atomic force microscopy. Electrochemistry Communications 2005, 7(6), 612–618. [Google Scholar]
- Kueng, A.; Kranz, C.; Lugstein, A.; Bertagnolli, E.; Mizaikoff, B. Integrated afm-secm in tapping mode: Simultaneous topographical and electrochemical imaging of enzyme activity. Angewandte Chemie-International Edition 2003, 42(28), 3238–3240. [Google Scholar]
- Kranz, C.; Kueng, A.; Lugstein, A.; Bertagnolli, E.; Mizaikoff, B. Mapping of enzyme activity by detection of enzymatic products during afm imaging with integrated secm-afm probes. Ultramicroscopy 2004, 100(3-4), 127–134. [Google Scholar]
- Jones, C.E.; Unwin, P. R.; Macpherson, J. V. In situ observation of the surface processes involved in dissolution from the cleavage surface of calcite in aqueous solution using combined scanning electrochemical-atomic force microscopy (secm-afm). Chemphyschem 2003, 4(2), 139–146. [Google Scholar]
- Kotitz, R.; Matz, H.; Trahms, L.; Koch, H.; Weitschies, W.; Rheinlander, T.; Semmler, W.; Bunte, T. Squid based remanence measurements for immunoassays. Ieee Transactions on Applied Superconductivity 1997, 7(2), 3678–3681. [Google Scholar]
- Matz, H.; Drung, D.; Hartwig, S.; Gross, H.; Kotitz, R.; Muller, W.; Vass, A.; Weitschies, W.; Trahms, L. A squid measurement system for immunoassays. Applied Superconductivity 1998, 6(10-12), 577–583. [Google Scholar]
- Kotitz, R.; Weitschies, W.; Trahms, L.; Brewer, W.; Semmler, W. Determination of the binding reaction between avidin and biotin by relaxation measurements of magnetic nanoparticles. Journal of Magnetism and Magnetic Materials 1999, 194(1-3), 62–68. [Google Scholar]
- Lange, J.; Kotitz, R.; Haller, A.; Trahms, L.; Semmler, W.; Weitschies, W. Magnetorelaxometry -a new binding specific detection method based on magnetic nanoparticles. Journal of Magnetism and Magnetic Materials 2002, 252(1-3), 381–383. [Google Scholar]
- Meyer, M.H. F.; Hartmann, M.; Krause, H. J.; Blankenstein, G.; Mueller-Chorus, B.; Oster, J.; Miethe, P.; Keusgen, M. Crp determination based on a novel magnetic biosensor. Biosensors & Bioelectronics 2007, 22(6), 973–979. [Google Scholar]
- Krause, H.-J.; Zhang, Y.; Wolters, N.; Miethe, P.; Plaksin, D. Method and device for selectively detecting ferromagnetic or superparamagnetic particles. 2003. [Google Scholar]
- Baselt, D.R.; Lee, G. U.; Colton, R. J. Biosensor based on force microscope technology. Journal of Vacuum Science & Technology B 1996, 14(2), 789–793. [Google Scholar]
- Larsson, K.; Kriz, K.; Kriz, D. Magnetic transducers in biosensors and bioassays. Analusis 1999, 27(7), 617–621. [Google Scholar]
- Kriz, C.B.; Radevik, K.; Kriz, D. Magnetic permeability measurements in bioanalysis and biosensors. Analytical Chemistry 1996, 68(11), 1966–1970. [Google Scholar]
- Babb, C.W.; Coon, D. R.; Rechnitz, G. A. Biomagnetic neurosensors .3. noninvasive sensors using magnetic stimulation and biomagnetic detection. Analytical Chemistry 1995, 67(4), 763–769. [Google Scholar]
- Baibich, M.N.; Broto, J. M.; Fert, A.; Vandau, F. N.; Petroff, F.; Eitenne, P.; Creuzet, G.; Friederich, A.; Chazelas, J. Giant magnetoresistance of (001)fe/(001) cr magnetic superlattices. Physical Review Letters 1988, 61(21), 2472–2475. [Google Scholar]
- Baselt, D.R.; Lee, G. U.; Natesan, M.; Metzger, S. W.; Sheehan, P. E.; Colton, R. J. A biosensor based on magnetoresistance technology. Biosensors & Bioelectronics 1998, 13(7-8), 731–739. [Google Scholar]
- Graham, D.L.; Ferreira, H. A.; Freitas, P. P. Magnetoresistive-based biosensors and biochips. Trends in Biotechnology 2004, 22(9), 455–462. [Google Scholar]
- Megens, M.; Prins, M. Magnetic biochips: a new option for sensitive diagnostics. Journal of Magnetism and Magnetic Materials 2005, 293(1), 702–708. [Google Scholar]
- Helali, S.; Martelet, C.; Abdelghani, A.; Maaref, M.A.; Jaffrezic-Renault, N. A disposable immunomagnetic electrochemical sensor based on functionalised magnetic beads on gold surface for the detection of atrazine. Electrochimica Acta 2006, 51(24), 5182–5186. [Google Scholar]
- Tang, D.P.; Yuan, R.; Chai, Y. Q. Direct electrochemical immunoassay based on immobilization of protein-magnetic nanoparticle composites on to magnetic electrode surfaces by sterically enhanced magnetic field force. Biotechnology Letters 2006, 28(8), 559–565. [Google Scholar]
- Rife, J.C.; Miller, M. M.; Sheehan, P. E.; Tamanaha, C. R.; Tondra, M.; Whitman, L. J. Design and performance of gmr sensors for the detection of magnetic microbeads in biosensors. Sensors and Actuators a-Physical 2003, 107(3), 209–218. [Google Scholar]
- Edelstein, R.L.; Tamanaha, C. R.; Sheehan, P. E.; Miller, M. M.; Baselt, D. R.; Whitman, L. J.; Colton, R. J. The barc biosensor applied to the detection of biological warfare agents. Biosensors & Bioelectronics 2000, 14(10-11), 805–813. [Google Scholar]
- Pavlickova, P.; Schneider, E.M.; Hug, H. Advances in recombinant antibody microarrays. Clinica Chimica Acta 2004, 343(1-2), 17–35. [Google Scholar]
- Lin, J.H.; Ju, H. X. Electrochemical and chemiluminescent immunosensors for tumor markers. Biosensors & Bioelectronics 2005, 20(8), 1461–1470. [Google Scholar]
- Dai, Z.; Yan, F.; Hua, Y.; Hu, X.Y.; Ju, H. X. Novel amperometric immunosensor for rapid separation-free immunoassay of carcinoembryonic antigen. Journal of Immunological Methods 2004, 287(1-2), 13–20. [Google Scholar]
- Yang, L.J.; Li, Y. B. Afm and impedance spectroscopy characterization of the immobilization of antibodies on indium-tin oxide electrode through self-assembled monolayer of epoxysilane and their capture of escherichia coli o157: H7. Biosensors & Bioelectronics 2005, 20(7), 1407–1416. [Google Scholar]
- Zhang, Y.Y.; Fung, Y. S.; Sun, H.; Zhu, D. R.; Yao, S. Z. Study of protein adsorption on polymer coatings surface by combining quartz crystal microbalance with electrochemical impedance methods. Sensors and Actuators B-Chemical 2005, 108(1-2), 933–942. [Google Scholar]
- Wink, T.; van Zuilen, S.; Bult, A.; van Bennekom, W. Self-assembled monolayers for biosensors. Analyst 1997, 122. [Google Scholar]
- Pan, D.W.; Chen, J. H.; Yao, S. Z.; Tao, W. Y.; Nie, L. H. An amperometric glucose biosensor based on glucose oxidase immobilized in electropolymerized poly(o-aminophenol) and carbon nanotubes composite film on a gold electrode. Analytical Sciences 2005, 21(4), 367–371. [Google Scholar]
- Sotiropoulou, S.; Gavalas, V.; Vamvakaki, V.; Chaniotakis, N.A. Novel carbon materials in biosensor systems. Biosensors & Bioelectronics 2003, 18(2-3), 211–215. [Google Scholar]
- Mena, M.L.; Yanez-Sedeno, P.; Pingarron, J. M. A comparison of different strategies for the construction of amperometric enzyme biosensors using gold nanoparticle-modified electrodes. Analytical Biochemistry 2005, 336(1), 20–27. [Google Scholar]
- Bauer, L.A.; Birenbaum, N. S.; Meyer, G. J. Biological applications of high aspect ratio nanoparticles. Journal of Materials Chemistry 2004, 14(4), 517–526. [Google Scholar]
- Yu, Y.Y.; Chang, S. S.; Lee, C. L.; Wang, C. R.C. Gold nanorods: Electrochemical synthesis and optical properties. Journal of Physical Chemistry B 1997, 101(34), 6661–6664. [Google Scholar]
- Yi, G.C.; Wang, C. R.; Park, W. I. Zno nanorods: synthesis, characterization and applications. Semiconductor Science and Technology 2005, 20(4), S22–S34. [Google Scholar]
- Kuznetsov, B.A.; Shumakovich, G. P.; Koroleva, O. V.; Yaropolov, A. I. On applicability of laccase as label in the mediated and mediatorless electroimmunoassay: effect of distance on the direct electron transfer between laccase and electrode. Biosensors & Bioelectronics 2001, 16(1-2), 73–84. [Google Scholar]
- Ghindilis, A.L.; Atanasov, P.; Wilkins, E. Enzyme-catalyzed direct electron transfer: Fundamentals and analytical applications. Electroanalysis 1997, 9(9), 661–674. [Google Scholar]
- Patolsky, F.; Weizmann, Y.; Willner, I. Long-range electrical contacting of redox enzymes by swcnt connectors. Angewandte Chemie-International Edition 2004, 43(16), 2113–2117. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell. 2002.
- Heller, A. Amperometric biosensors. Current Opinion in Biotechnology 1996, 7(1), 50–54. [Google Scholar]
- Azevedo, A.; Martins, V.; Prazeres, D.; Vojinovic, V.; Cabral, J.; Fonseca, L. Horseradish peroxidase: a valuable tool in biotechnology. Biotechnology annual review 2003, 9, 199–247. [Google Scholar]
- Zhen, G.L.; Eggli, V.; Voros, J.; Zammaretti, P.; Textor, M.; Glockshuber, R.; Kuennemann, E. Immobilization of the enzyme beta-lactamase on biotin-derivatized poly(l-lysine)-g-poly(ethylene glycol)-coated sensor chips: A study on oriented attachment and surface activity by enzyme kinetics and in situ optical sensing. Langmuir 2004, 20(24), 10464–10473. [Google Scholar]
- Chaniotakis, N.A. Enzyme stabilization strategies based on electrolytes and polyelectrolytes for biosensor applications. Analytical and Bioanalytical Chemistry 2004, 378(1), 89–95. [Google Scholar]
- O'Fagain, C. Enzyme stabilization - recent experimental progress. Enzyme and Microbial Technology 2003, 33(2-3), 137–149. [Google Scholar]
- Grieshaber, D.; Reimhult, E.; Vrs, J. Enzymatic biosensors towards a multiplexed electronic detection system for early cancer diagnostics. In 2nd IEEE -NEMS; volume 1, pp. 402–405. Bangkok, 2007. [Google Scholar]
- Sotiropoulou, S.; Vamvakaki, V.; Chaniotakis, N.A. Stabilization of enzymes in nanoporous materials for biosensor applications. Biosensors & Bioelectronics 2005, 20(8), 1674–1679. [Google Scholar]
- Sung, W.J.; Bae, Y. H. Glucose oxidase, lactate oxidase, and galactose oxidase enzyme electrode based on polypyrrole with polyanion/peg/enzyme conjugate dopant. Sensors and Actuators B-Chemical 2006, 114(1), 164–169. [Google Scholar]
- Kim, J.; Grate, J.W.; Wang, P. Nanostructures for enzyme stabilization. Chemical Engineering Science 2006, 61(3), 1017–1026. [Google Scholar]
- Lu, B.; Smyth, M.R.; OKennedy, R. Oriented immobilization of antibodies and its applications in immunoassays and immunosensors. Analyst 1996, 121(3), R29–R32. [Google Scholar]
- LaGraff, J.R.; Chu-LaGraff, Q. Scanning force microscopy and fluorescence microscopy of microcontact printed antibodies and antibody fragments. Langmuir 2006, 22(10), 4685–4693. [Google Scholar]
- Wacker, R.; Schroder, H.; Niemeyer, C.M. Performance of antibody microarrays fabricated by either dna-directed immobilization, direct spotting, or streptavidin-biotin attachment: a comparative study. Analytical Biochemistry 2004, 330(2), 281–287. [Google Scholar]
- Peluso, P.; Wilson, D.S.; Do, D.; Tran, H.; Venkatasubbaiah, M.; Quincy, D.; Heidecker, B.; Poindexter, K.; Tolani, N.; Phelan, M.; Witte, K.; Jung, L. S.; Wagner, P.; Nock, S. Optimizing antibody immobilization strategies for the construction of protein microarrays. Analytical Biochemistry 2003, 312(2), 113–124. [Google Scholar]
- Padeste, C.; Grubelnik, A.; Steiger, B.; Hefti, J.; Tiefenauer, L. Molecular architectures for enzyme sensors. PSI annual report 2001 2001. [Google Scholar]
- Zacco, E.; Pividori, M.I.; Alegret, S. Electrochemical biosensing based on universal affinity biocomposite platforms. Biosensors & Bioelectronics 2006, 21(7), 1291–1301. [Google Scholar]
- Danczyk, R.; Krieder, B.; North, A.; Webster, T.; HogenEsch, H.; Rundell, A. Comparison of antibody functionality using different immobilization methods. Biotechnology and Bioengineering 2003, 84(2), 215–223. [Google Scholar]
- Nielsen, U.B.; Geierstanger, B. H. Multiplexed sandwich assays in microarray format. Journal of Immunological Methods 2004, 290(1-2), 107–120. [Google Scholar]
- Sarkar, P.; Pal, P.S.; Ghosh, D.; Setford, S. J.; Tothill, I. E. Amperometric biosensors for detection of the prostate cancer marker (psa). International Journal of Pharmaceutics 2002, 238(1-2), 1–9. [Google Scholar]
- Haab, B.B. Applications of antibody array platforms. Current Opinion in Biotechnology 2006, 17(4), 415–421. [Google Scholar]
- Peterson, J.; Green, G.; Iida, K.; Caldwell, B.; Kerrison, P.; Bernich, S.; Aoyagi, K.; Lee, S.R. Detection of hepatitis c core antigen in the antibody negative ‘window’ phase of hepatitis c infection. Vox Sanguinis 2000, 78(2), 80–85. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nature Biotechnology 2005, 23(9), 1126–1136. [Google Scholar]
- Hamerscasterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally-occurring antibodies devoid of light-chains. Nature 1993, 363(6428), 446–448. [Google Scholar]
- Vikholm, I. Self-assembly of antibody fragments and polymers onto gold for immunosensing. Sensors and Actuators B-Chemical 2005, 106(1), 311–316. [Google Scholar]
- Ellington, A.D.; Szostak, J. W. Invitro selection of rna molecules that bind specific ligands. Nature 1990, 346(6287), 818–822. [Google Scholar]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment - rna ligands to bacteriophage-t4 dna-polymerase. Science 1990, 249(4968), 505–510. [Google Scholar]
- Tombelli, S.; Minunni, A.; Mascini, A. Analytical applications of aptamers. Biosensors & Bioelectronics 2005, 20(12), 2424–2434. [Google Scholar]
- O'Sullivan, C.K. Aptasensors - the future of biosensing. Analytical and Bioanalytical Chemistry 2002, 372(1), 44–48. [Google Scholar]
- Caruso, F.; Caruso, R.A.; Mohwald, H. Nanoengineering of inorganic and hybrid hollow spheres by colloidal templating. Science 1998, 282(5391), 1111–1114. [Google Scholar]
- Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davis, S. A.; Mohwald, H. Novel hollow polymer shells by colloid-templated assembly of polyelectrolytes. Angewandte Chemie-International Edition 1998, 37(16), 2202–2205. [Google Scholar]
- Antipov, A.A.; Sukhorukov, G. B. Polyelectrolyte multilayer capsules as vehicles with tunable permeability. Advances in Colloid and Interface Science 2004, 111(1-2), 49–61. [Google Scholar]
- Heuberger, R.; Sukhorukov, G.; Voros, J.; Textor, M.; Mohwald, H. Biofunctional polyelectrolyte multilayers and microcapsules: Control of non-specific and bio-specific protein adsorption. Advanced Functional Materials 2005, 15(3), 357–366. [Google Scholar]
- Gao, C. Y.; Liu, X. Y.; Shen, J. C.; Mohwald, H. Spontaneous deposition of horseradish peroxidase into polyelectrolyte multilayer capsules to improve its activity and stability. Chemical Communications 2002, (17), 1928–1929. [Google Scholar]
- Volodkin, D.V.; Petrov, A. I.; Prevot, M.; Sukhorukov, G. B. Matrix polyelectrolyte microcapsules: New system for macromolecule encapsulation. Langmuir 2004, 20(8), 3398–3406. [Google Scholar]
- Walde, P.; Ichikawa, S. Enzymes inside lipid vesicles: Preparation, reactivity and applications. Biomolecular Engineering 2001, 18(4), 143–177. [Google Scholar]
- Nasseau, M.; Boublik, Y.; Meier, W.; Winterhalter, M.; Fournier, D. Substrate-permeable encapsulation of enzymes maintains effective activity, stabilizes against denaturation, and protects against proteolytic degradation. Biotechnology and Bioengineering 2001, 75(5), 615–618. [Google Scholar]
- Winterhalter, M.; Hilty, C.; Bezrukov, S.M.; Nardin, C.; Meier, W.; Fournier, D. Controlling membrane permeability with bacterial porins: application to encapsulated enzymes. Talanta 2001, 55(5), 965–971. [Google Scholar]
- Hill, K.J.; Kaszuba, M.; Creeth, J. E.; Jones, M. N. Reactive liposomes encapsulating a glucose oxidase-peroxidase system with antibacterial activity. Biochimica Et Biophysica Acta-Biomembranes 1997, 1326(1), 37–46. [Google Scholar]
- Torchilin, V.P. Peg-based micelles as carriers of contrast agents for different imaging modalities. Advanced Drug Delivery Reviews 2002, 54(2), 235–252. [Google Scholar]
- Torchilin, V.P. Targeted polymeric micelles for delivery of poorly soluble drugs. Cellular and Molecular Life Sciences 2004, 61, 2549–2559. [Google Scholar]
- Gaucher, G.; Dufresne, M.H.; Sant, V. P.; Kang, N.; Maysinger, D.; Leroux, J. C. Block copolymer micelles: preparation, characterization and application in drug delivery. Journal of Controlled Release 2005, 109(1-3), 169–188. [Google Scholar]
- Thurmond, K.B.; Huang, H. Y.; Clark, C. G.; Kowalewski, T.; Wooley, K. L. Shell cross-linked polymer micelles: stabilized assemblies with great versatility and potential. Colloids and Surfaces B-Biointerfaces 1999, 16(1-4), 45–54. [Google Scholar]
- Harada, A.; Kataoka, K. Novel polyion complex micelles entrapping enzyme molecules in the core: Preparation of narrowly-distributed micelles from lysozyme and poly(ethylene glycol)-poly(aspartic acid) block copolymer in aqueous medium. Macromolecules 1998, 31(2), 288–294. [Google Scholar]
- Harada, A.; Kataoka, K. Pronounced activity of enzymes through the incorporation into the core of polyion complex micelles made from charged block copolymers. Journal of Controlled Release 2001, 72(1-3), 85–91. [Google Scholar]
- Heller, A. Electrical connection of enzyme redox centers to electrodes. Journal of Physical Chemistry 1992, 96(9), 3579–3587. [Google Scholar]
- Dong, S.J.; Guo, Y. Z. Organic-phase enzyme electrode operated in water-free solvents. Analytical Chemistry 1994, 66(22), 3895–3899. [Google Scholar]
- Mano, N.; Mao, F.; Heller, A. On the parameters affecting the characteristics of the “wired” glucose oxidase anode. Journal of Electroanalytical Chemistry 2005, 574(2), 347–357. [Google Scholar]
- Mitala, J.J.; Michael, A. C. Improving the performance of electrochemical microsensors based on enzymes entrapped in a redox hydrogel. Analytica Chimica Acta 2006, 556(2), 326–332. [Google Scholar]
- Mao, F.; Mano, N.; Heller, A. Long tethers binding redox centers to polymer backbones enhance electron transport in enzyme “wiring” hydrogels. Journal of the American Chemical Society 2003, 125(16), 4951–4957. [Google Scholar]
- Braun, S.; Rappoport, S.; Zusman, R.; Avnir, D.; Ottolenghi, M. Biochemically active sol-gel glasses - the trapping of enzymes. Materials Letters 1990, 10(1-2), 1–5. [Google Scholar]
- Avnir, D.; Coradin, T.; Lev, O.; Livage, J. Recent bio-applications of sol-gel materials. Journal of Materials Chemistry 2006, 16(11), 1013–1030. [Google Scholar]
- Jin, W.; Brennan, J.D. Properties and applications of proteins encapsulated within sol-gel derived materials. Analytica Chimica Acta 2002, 461(1), 1–36. [Google Scholar]
- Lei, C.X.; Hu, S. Q.; Gao, N.; Shen, G. L.; Yu, R. Q. An amperometric hydrogen peroxide biosensor based on immobilizing horseradish peroxidase to a nano-au monolayer supported by sol-gel derived carbon ceramic electrode. Bioelectrochemistry 2004, 65(1), 33–39. [Google Scholar]
- Trojanowicz, M. Miniaturized biochemical sensing devices based on planar bilayer lipid membranes. Fresenius Journal of Analytical Chemistry 2001, 371(2), 246–260. [Google Scholar]
- Xu, J.; Wang, X.B.; Ensign, B.; Li, M.; Wu, L.; Guia, A.; Xu, J. Q. Ion-channel assay technologies: quo vadis? Drug Discovery Today 2001, 6(24), 1278–1287. [Google Scholar]
- Howard, A.D.; McAllister, G.; Feighner, S. D.; Liu, Q. Y.; Nargund, R. P.; Van der Ploeg, L. H.T.; Patchett, A. A. Orphan g-protein-coupled receptors and natural ligand discovery. Trends in Pharmacological Sciences 2001, 22(3), 132–140. [Google Scholar]
- Schmitt, E.; Nurbani, M.; Bushby, R.; Steinem, C. Electrically insulating pore-suspending membranes on highly ordered porous alumina obtained from vesicle spreading. Soft Matter 2008. [Google Scholar]
- Han, X.; DiBernardino, M.; Studer, A.; Sehr, H.; Geissbhler, I.; Winkler, F.; Tiefenauer, L. Nanopore arrays for stable and functional free-standing lipid bilayers. Advanced Materials 2007, 19, 4466–4470. [Google Scholar]
- Reimhult, E.; Kumar, K. Membrane biosensor platforms using nano- and mocroporous supports. Trends in Biotechnology 2008, 26(2), 82–89. [Google Scholar]
- Castellana, E.T.; Cremer, P. S. Solid supported lipid bilayers: From biophysical studies to sensor design. Surface Science Reports 2006, 61(10), 429–444. [Google Scholar]
- Ide, T.; Ichikawa, T. A novel method for artificial lipid-bilayer formation. Biosensors & Bioelectronics 2005, 21(4), 672–677. [Google Scholar]
- Janshoff, A.; Steinem, C. Transport across artificial membranes - an analytical perspective. Analytical and Bioanalytical Chemistry 2006, 385(3), 433–451. [Google Scholar]
- Sackmann, E. Supported membranes: Scientific and practical applications. Science 1996, 271(5245), 43–48. [Google Scholar]
- Lang, H.; Duschl, C.; Vogel, H. A new class of thiolipids for the attachment of lipid bilayers on gold surfaces. Langmuir 1994, 10(1), 197–210. [Google Scholar]
- Naumann, R.; Schiller, S.M.; Giess, F.; Grohe, B.; Hartman, K. B.; Karcher, I.; Koper, I.; Lubben, J.; Vasilev, K.; Knoll, W. Tethered lipid bilayers on ultraflat gold surfaces. Langmuir 2003, 19(13), 5435–5443. [Google Scholar]
- Naumann, R.; Walz, D.; Schiller, S.M.; Knoll, W. Kinetics of valinomycin-mediated k+ ion transport through tethered bilayer lipid membranes. Journal of Electroanalytical Chemistry 2003, 550, 241–252. [Google Scholar]
- Steinem, C.; Galla, H.J.; Janshoff, A. Interaction of melittin with solid supported membranes. Physical Chemistry Chemical Physics 2000, 2(20), 4580–4585. [Google Scholar]
- Steinem, C.; Janshoff, A.; Galla, H.J.; Sieber, M. Impedance analysis of ion transport through gramicidin channels incorporated in solid supported lipid bilayers. Bioelectrochemistry and Bioenergetics 1997, 42(2), 213–220. [Google Scholar]
- Gritsch, S.; Nollert, P.; Jahnig, F.; Sackmann, E. Impedance spectroscopy of porin and gramicidin pores reconstituted into supported lipid bilayers on indium-tin-oxide electrodes. Langmuir 1998, 14(11), 3118–3125. [Google Scholar]
- Yin, P.; Burns, C.J.; Osman, P. D.J.; Cornell, B. A. A tethered bilayer sensor containing alamethicin channels and its detection of amiloride based inhibitors. Biosensors & Bioelectronics 2003, 18(4), 389–397. [Google Scholar]
- Glazier, S.A.; Vanderah, D. J.; Plant, A. L.; Bayley, H.; Valincius, G.; Kasianowicz, J. J. Reconstitution of the pore-forming toxin alpha-hemolysin in phospholipid/18-octadecyl-1-thiahexa(ethylene oxide) and phospholipid/n-octadecanethiol supported bilayer membranes. Langmuir 2000, 16(26), 10428–10435. [Google Scholar]
- Hirano, A.; Wakabayashi, M.; Matsuno, Y.; Sugawara, M. A single-channel sensor based on gramicidin controlled by molecular recognition at bilayer lipid membranes containing receptor. Biosensors & Bioelectronics 2003, 18(8), 973–983. [Google Scholar]
- Knapp, O.; Benz, R.; Gibert, M.; Marvaud, J.C.; Popoff, M. R. Interaction of clostridium perfringens iota-toxin with lipid bilayer membranes - demonstration of channel formation by the activated binding component ib and channel block by the enzyme component ia. Journal of Biological Chemistry 2002, 277(8), 6143–6152. [Google Scholar]
- Bayley, H.; Jayasinghe, L. Functional engineered channels and pores - (review). Molecular Membrane Biology 2004, 21(4), 209–220. [Google Scholar]
- Ataka, K.; Giess, F.; Knoll, W.; Naumann, R.; Haber-Pohlmeier, S.; Richter, B.; Heberle, J. Oriented attachment and membrane reconstitution of his-tagged cytochrome c oxidase to a gold electrode: In situ monitoring by surface-enhanced infrared absorption spectroscopy. Journal of the American Chemical Society 2004, 126(49), 16199–16206. [Google Scholar]
- Naumann, R.; Baumgart, T.; Graber, P.; Jonczyk, A.; Offenhausser, A.; Knoll, W. Proton transport through a peptide-tethered bilayer lipid membrane by the h+-atp synthase from chloroplasts measured by impedance spectroscopy. Biosensors & Bioelectronics 2002, 17(1-2), 25–34. [Google Scholar]
- Ottenbacher, D.; Kindervater, R.; Gimmel, P.; Klee, B.; Jahnig, F.; Gopel, W. Developing biosensors with ph-isfet transducers utilizing lipid bilayer-membranes with transport proteins. Sensors and Actuators B-Chemical 1992, 6(1-3), 192–196. [Google Scholar]
- Jeuken, L.J. C.; Connell, S. D.; Nurnabi, M.; O'Reilly, J.; Henderson, P. J.F.; Evans, S. D.; Bushby, R. J. Direct electrochemical interaction between a modified gold electrode and a bacterial membrane extract. Langmuir 2005, 21(4), 1481–1488. [Google Scholar]
- Doyle, D.A.; Cabral, J. M.; Pfuetzner, R. A.; Kuo, A. L.; Gulbis, J. M.; Cohen, S. L.; Chait, B. T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of k+ conduction and selectivity. Science 1998, 280(5360), 69–77. [Google Scholar]
- Andersson, M.; Keizer, H.M.; Zhu, C. Y.; Fine, D.; Dodabalapur, A.; Duran, R. S. Detection of single ion channel activity on a chip using tethered bilayer membranes. Langmuir 2007, 23(6), 2924–2927. [Google Scholar]
- Cornell, B.A.; BraachMaksvytis, V. L.B.; King, L. G.; Osman, P. D.J.; Raguse, B.; Wieczorek, L.; Pace, R. J. A biosensor that uses ion-channel switches. Nature 1997, 387(6633), 580–583. [Google Scholar]
- Woodhouse, G.; King, L.; Wieczorek, L.; Osman, P.; Cornell, B. The ion channel switch biosensor. Journal of Molecular Recognition 1999, 12(5), 328–334. [Google Scholar]
- Nikolelis, D.P.; Siontorou, C. G.; Krull, U. J.; Katrivanos, P. L. Ammonium ion minisensors from self-assembled bilayer lipid membranes using gramicidin as an ionophore. modulation of ammonium selectivity by platelet-activating factor. Analytical Chemistry 1996, 68(10), 1735–1741. [Google Scholar]
- Blake, S.; Mayer, T.; Mayer, M.; Yang, J. Monitoring chemical reactions by using ion-channel-forming peptides. Chembiochem 2006, 7(3), 433–435. [Google Scholar]
- Stora, T.; Lakey, J.H.; Vogel, H. Ion-channel gating in transmembrane receptor proteins: Functional activity in tethered lipid membranes. Angewandte Chemie-International Edition 1999, 38(3), 389–392. [Google Scholar]
- Boulmedais, F.; Frisch, B.; Etienne, O.; Lavalle, P.; Picart, C.; Ogier, J.; Voegel, J.C.; Schaaf, P.; Egles, C. Polyelectrolyte multilayer films with pegylated polypeptides as a new type of anti-microbial protection for biomaterials. Biomaterials 2004, 25(11), 2003–2011. [Google Scholar]
- Templin, M.F.; Stoll, D.; Schrenk, M.; Traub, P. C.; Vohringer, C. F.; Joos, T. O. Protein microarray technology. Trends in Biotechnology 2002, 20(4), 160–166. [Google Scholar]
- Haab, B.B. Antibody arrays in cancer research. Molecular & Cellular Proteomics 2005, 4(4), 377–383. [Google Scholar]
- Romer, W.; Lam, Y.H.; Fischer, D.; Watts, A.; Fischer, W. B.; Goring, P.; Wehrspohn, R. B.; Gosele, U.; Steinem, C. Channel activity of a viral transmembrane peptide in micro-blms: Vpu(1-32) from hiv-1. Journal of the American Chemical Society 2004, 126(49), 16267–16274. [Google Scholar]
- Danelon, C.; Perez, J.B.; Santschi, C.; Brugger, J.; Vogel, H. Cell membranes suspended across nanoaperture arrays. Langmuir 2006, 22(1), 22–25. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}























© 2008 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Grieshaber, D.; MacKenzie, R.; Vörös, J.; Reimhult, E. Electrochemical Biosensors - Sensor Principles and Architectures. Sensors 2008, 8, 1400-1458. https://doi.org/10.3390/s80314000
Grieshaber D, MacKenzie R, Vörös J, Reimhult E. Electrochemical Biosensors - Sensor Principles and Architectures. Sensors. 2008; 8(3):1400-1458. https://doi.org/10.3390/s80314000
Chicago/Turabian StyleGrieshaber, Dorothee, Robert MacKenzie, Janos Vörös, and Erik Reimhult. 2008. "Electrochemical Biosensors - Sensor Principles and Architectures" Sensors 8, no. 3: 1400-1458. https://doi.org/10.3390/s80314000
APA StyleGrieshaber, D., MacKenzie, R., Vörös, J., & Reimhult, E. (2008). Electrochemical Biosensors - Sensor Principles and Architectures. Sensors, 8(3), 1400-1458. https://doi.org/10.3390/s80314000