1. Introduction
Lipolytic enzymes are one of the most important components of the biochemical processes. At the same time, triacylglycerol acylhydrolases (EC 3.1.1.3) that hydrolyze triacylglycerols at the oil/water interface have wide applications as detergent additives, digestive aids, as well as in the paper and food industries [
1-
3]. Unlike other bond-cleaving enzymes, e.g., proteases, hydrolysis by lipases is carried out in heterogeneous multiphase systems. In many cases, the environment of the enzyme at the substrate/liquid interface plays an important role for the overall enzymatic activity of these proteins [
1-
4]. Thus, the ability to monitor enzymatic activity of lipases under these conditions is of paramount importance.
Recently, a novel electrochemical technique for the assay of lipase activity has been described [
5]. The method utilizes a solid supported lipase substrate, which is formed by dripping and drying a small amount of an ethanol solution of 9-(5′-ferrocenylpentanoyloxy)nonyl disulfide (FPONDS; [Fc-(CH
2)
4COO(CH
2)
9S-]
2, where Fc is the ferrocene) on the gold electrode surface modified by a hexanethiol self-assembled monolayer. The redox-active ferrocene group of FPONDS generates the amperometric signal, the intensity of which is proportional to the number of FPONDS molecules at the interface. Electrochemical and surface-enhanced infrared absorption spectroscopic data, as well as control experiments with an engineered, deactivated mutant enzyme, have demonstrated that the wild-type lipase from
Thermomyces lanuginosus (TLL) is capable of cleaving the ester bonds of FPONDS molecules via an enzymatic hydrolysis mechanism, which includes the adsorption of the lipase onto the substrate surface. The interfacial enzymatic process liberates ferrocene groups from the electrode surface triggering a decay of the electrochemical signal. The rate of the electrochemical signal decrease is proportional to the lipase activity.
However, in exclusively experimental work [
5], no kinetic model has been proposed to account for the features of amperometric biosensor response, namely, current decay vs. time upon enzyme action. This paper is intended to fill this gap.
2. Kinetic model
This paper analyzes bioelectroanalytical system that is significantly different from recently discussed amperometric system of lipase activity determination [
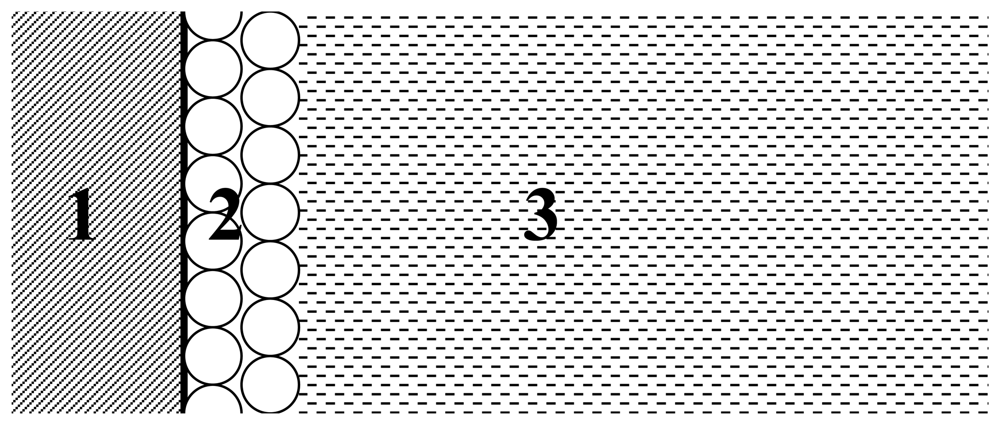
6], where enzyme acts on the surface of substrate-bearing micelles spread in the solution. Currently modeled system is schematically presented in
Fig. 1.
The processes that occur at the interface of zones 2 and 3 could be described in the following schematic form which is most commonly used for the description of lipase interfacial activation [
7]:
where E is the enzyme in solution, E * is the enzyme attached to the surface of substrate (at the interface of zones 2 and 3 in
Fig. 1), U is the ferrocene-based substrate FPONDS substrate on the gold electrode surface, E*U is the enzyme-substrate complex, and P represents the reaction product. The change of U concentration as a function time is the object of our computational simulations as it is directly proportional to experimentally registered electrode signal (see, for instance,
Fig. 1 in Ref.
5).
It is assumed that lipase solution is distributed evenly and its diffusion could be not taken into account. It is also assumed that the redox-active reaction product (ferrocene-based) leaves sensor surface quite fast and its diffusion could be estimated as instantaneous. The system under discussion can be described by classical mathematical model of reaction kinetics:
where symbols in italics
E,
E*,
E*
U,
P and
U represent concentrations;
S is the interfacial area of electrode;
V is the total volume of solution;
kp is the rate constant of enzyme adsorption at the electrode surface,
kD is the enzyme desorption rate constant,
k1 is the rate constant of enzyme-substrate complex (E*U) formation,
k-1 is the rate constant of E*U dissociation,
k2 is the catalytic rate constant of enzymatic reaction, and
t is time.
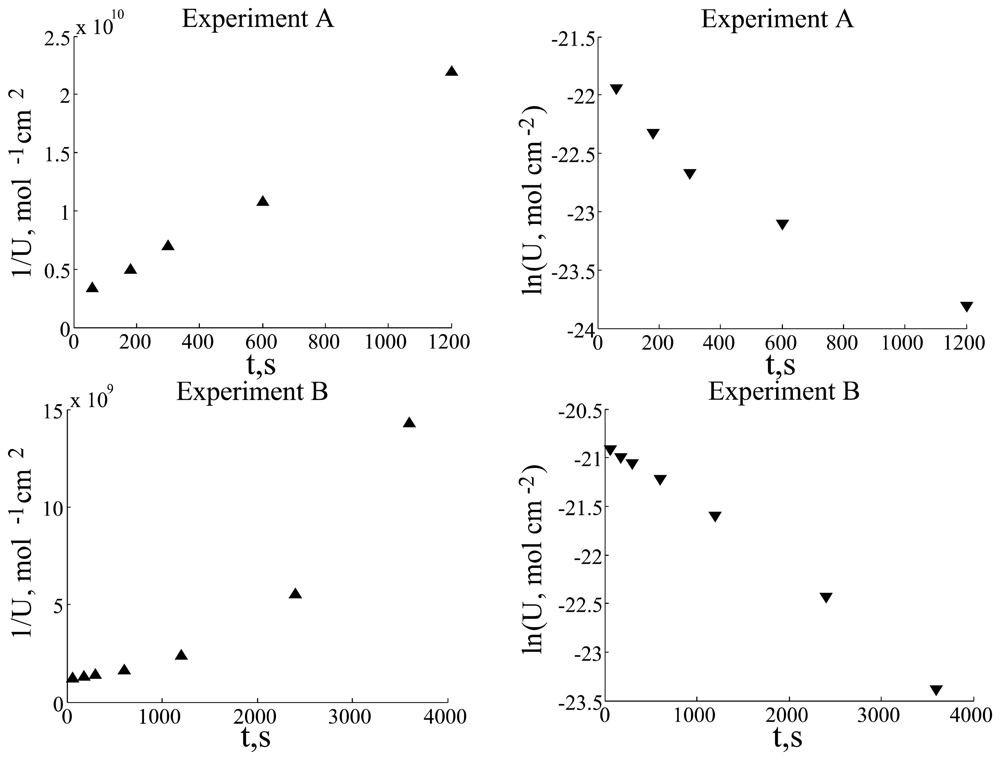
This model allowed good fitting only for a part of experimental data available (data not shown), which had strongly expressed exponential character of substrate concentration decrease (
Fig. 2, experiment B; for the characteristics of different experiments, see parameters in the table).
However, another part of experimental data exhibited
U decrease asymptotically proportional to t
-1 (
Fig 2, experiment A). Thus, the model of
Eq. (3) was modified by adding a non-linear term of substrate wash off from the electrode surface, which allowed much better fitting results. Here, it should be noted that in work [
5] the wash off effect of substrate has been observed experimentally in the solutions without added enzyme (see
Fig. 1 in paper [
5]). Therefore, we have reasonable grounds to believe that this process also occurs in the solutions containing enzyme.
Thus, slightly modified system of non-linear differential equations can be written by
Eq. (4):
where definitions are the same as for
Eq. (3), and
ku is the substrate wash off rate constant and
U0 is the initial substrate concentration on the electrode surface.
Non-linear wash off term is quite unusual, but it could be explained in a simplified way as complex outcome of two different linear wash off rates: one for the electrode surface/substrate boundary (stronger bond, lower wash off rate) and second (weaker attraction, much higher wash off rate) for, say, substrate/substrate boundary. It is possible that during the process of modified electrode preparation substrate forms only very few substrate/substrate boundaries (pseudo-multilayer interfacial structure). Thus, initially wash off rate could be seen as linearly (in respect to the substrate concentration) dropping from high value for the substrate/substrate boundary, down to low value for the electrode/substrate boundary, and the whole process then becomes second order with respect to the substrate concentration. By way of illustration, let's assume that the wash-off rate constant (k) changes linearly with relative substrate concentration: k=a·U/U0+b, where a and b are the constants, so non-linearity could be introduced by substituting the wash-off rate constant in standard linear wash-off model: dU/dt = -kU.
3. Computer simulation setup and results
The series of computational simulations were performed to investigate how electrode readings would differ if this amperometric biosensor worked under presented model and how they would match experimental data (experimental results were obtained as described in [
5], converting the integrated electrode peak current of the FPONDS-modified electrode to the surfaces concentration of ferrocene functional groups). The following values were used in our calculations:
V = 4 cm
3,
k2 = 75 s
-1,
k-1 = 10 s
-1,
kp = 100 cm s
-1,
kD = 0.025 s
-1,
SA = 5.07×10
-2 cm
2,
SB = 5.19×10
-2 cm
2,
SC = 5.23×10
-2 cm,
SD=5.23×10
-2 cm
2. The values of four kinetic constants selected as a starting point for modeling were the same as in paper [
6]. Besides, the following initial conditions were applied:
E(0) =
E0,
E*(0) =
E*
S(0) =
P(0) = 0,
U(0) =
U0. The values of initial
E0 and
U0 concentrations varied from experiment to experiment,
k1 and
ku were subject of change for achieving better fitting (weighted least squares method was used) between experimental and simulation data. Non-linear ordinary equation system (4) was solved using Matlab (Matlab Release 14, The MathWorks Inc., Natick, USA) ODE solver for stiff problems. Solution time interval was 0..6000 seconds. The initial concentrations and best-fitted constants are presented in the
Table 1.
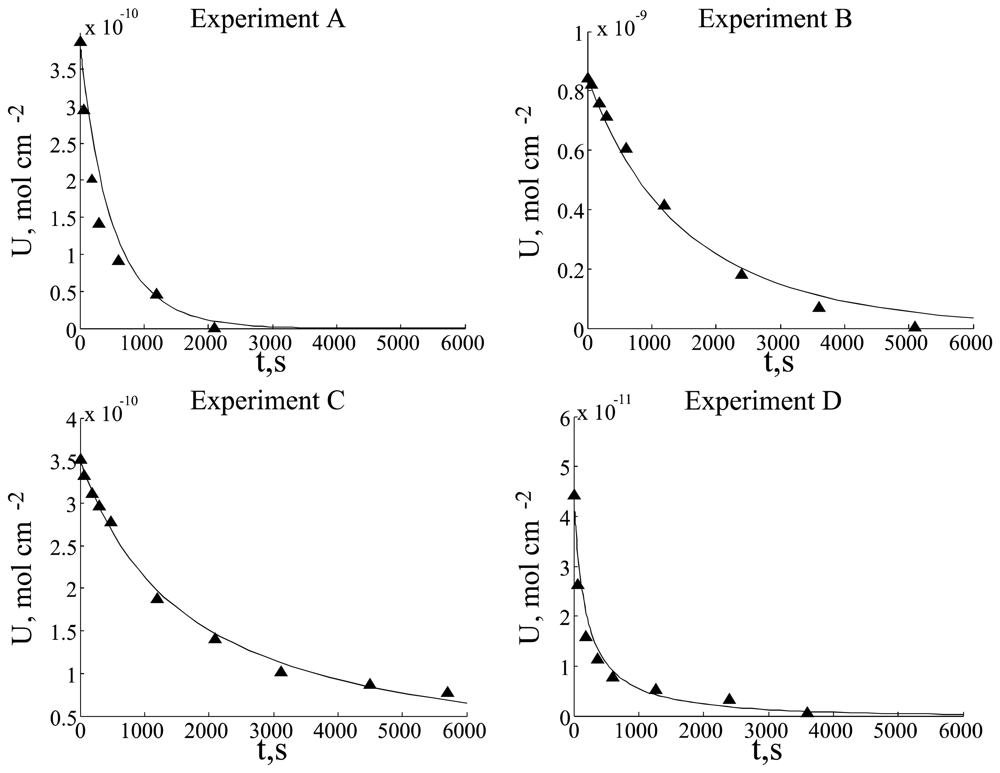
Experimental data and simulation results are presented in
Fig. 3.
Experimental data were analyzed as logarithmic and t-1 graphs. These graphs reveal that experiments A, C and D strongly exhibit inverse dependence on time, whereas the data of experiment B has more exponential character. Such different graph characters could be explained as two term competition in dU/dt differential equation: first and second order (in respect to the substrate concentration) terms. First order term predominated over the second order term in experiment B, but second order term predominated over the first order term in experiments A, C and D. These observations enabled us to improve the model and to get better fitting between simulation and experimental data.
Finally, it is worth noting that the values of kinetic constant
k1 obtained in this work are lower by ca. three orders of magnitude compared to the value reported in our earlier study [
6]. Most likely, the difference is determined by different chemical nature of substrate head-groups in work [
6] (dicyanohydroquinone-based group) and the present study (ferrocene-based group), since
k1 reflects molecular event of substrate binding in the enzyme active center.





{kind=link}
{kind=link}
{kind=link}