Over-the-Counter Biosensors: Past, Present, and Future

Abstract
:1. Introduction
2. Metabolite Sensors
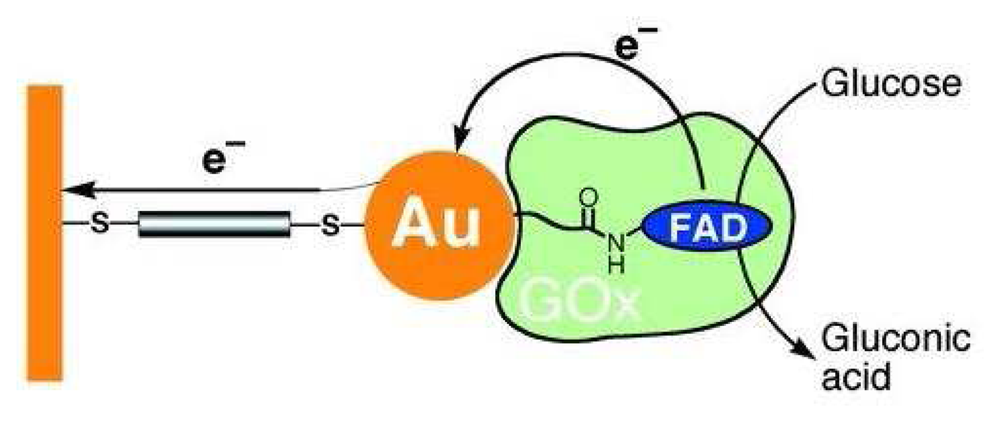
2.1. Basic Principles of Electrochemical Glucose Biosensors
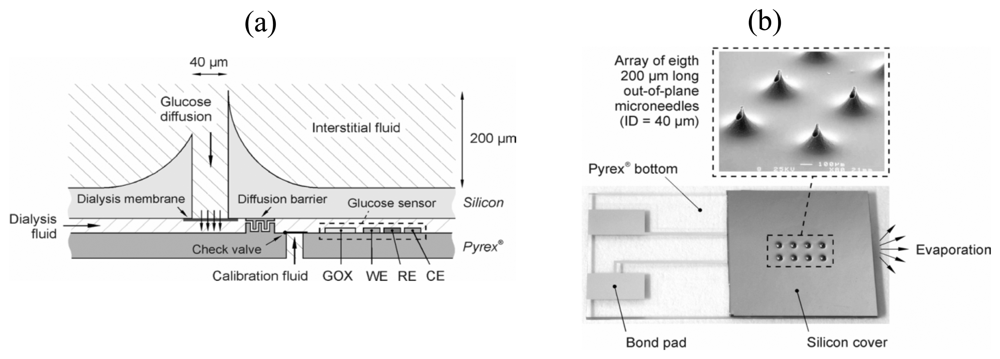
2.2. Types of Glucose Meters
2.3. Current Glucose Meters Market Situation
2.4. Future Prospects of Metabolite Biosensors
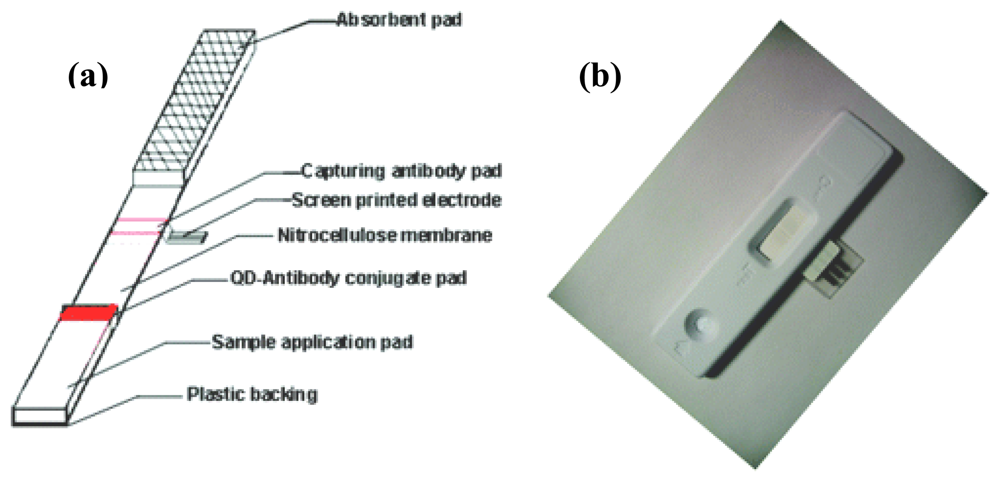
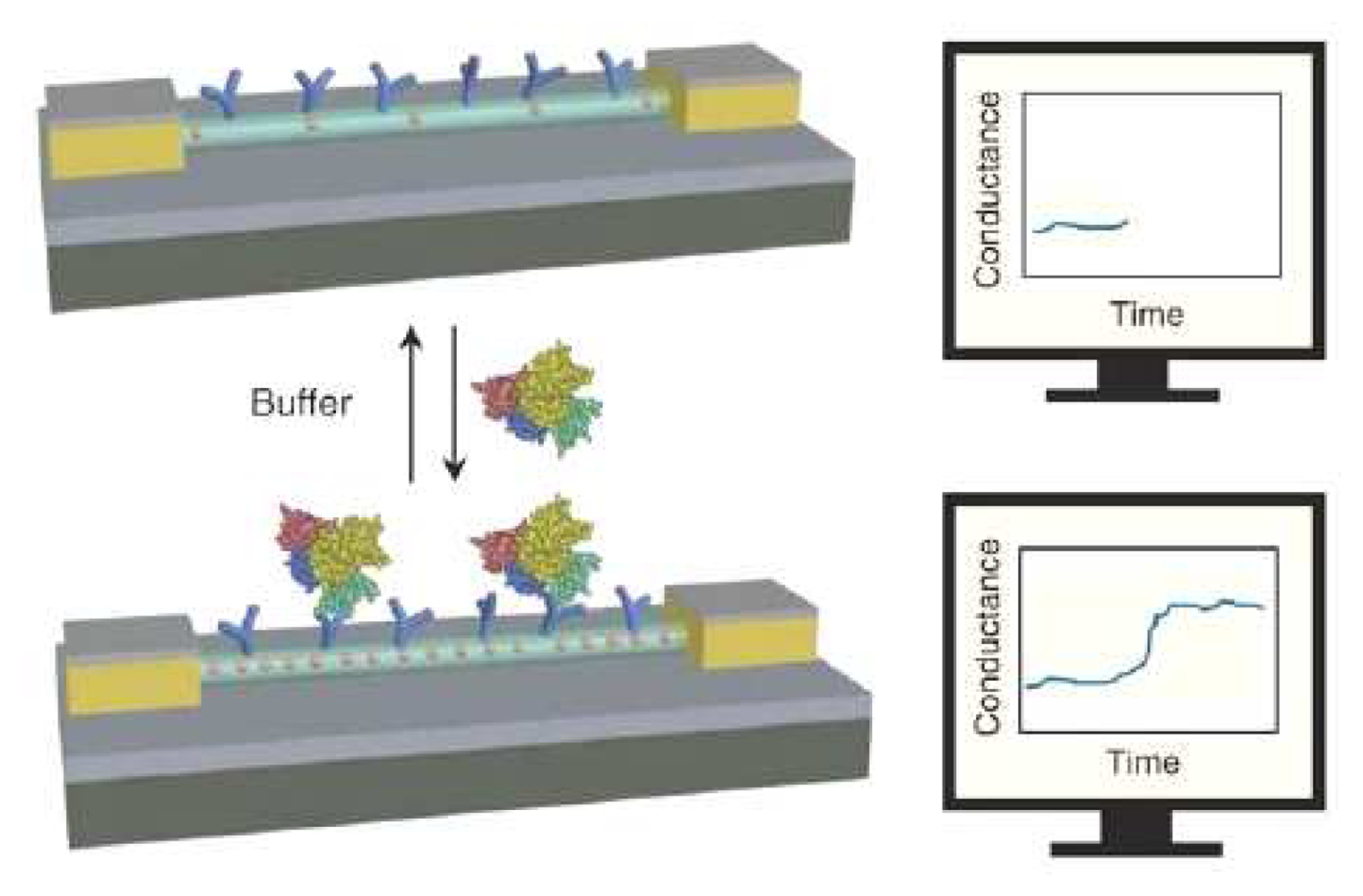
3. Protein Sensors
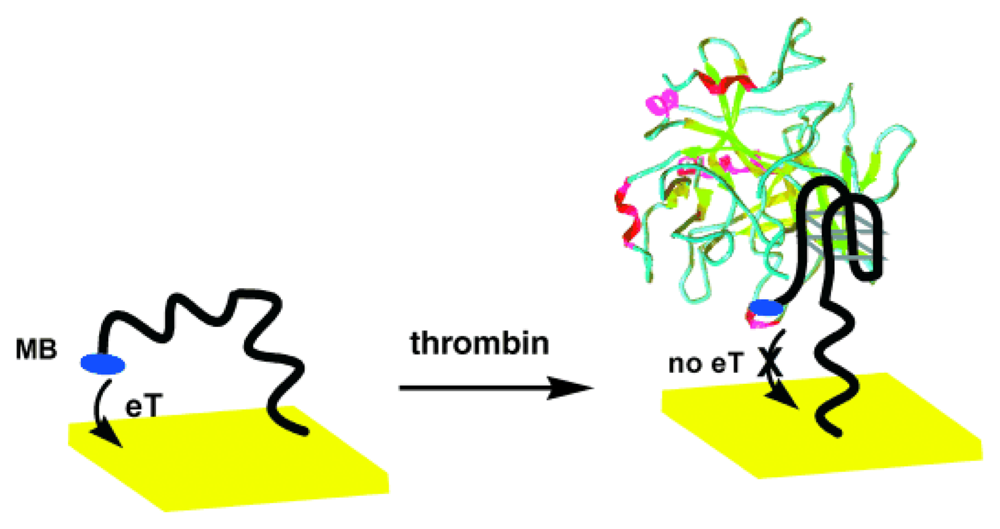
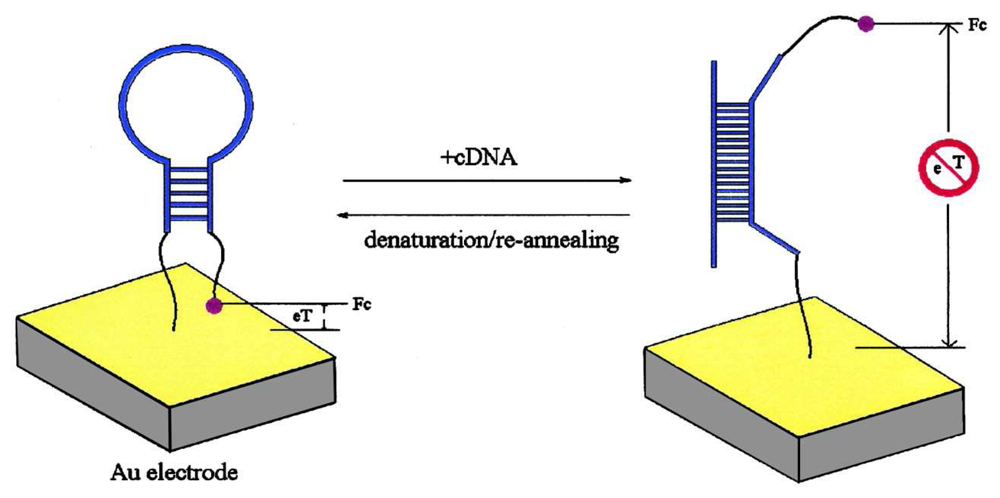
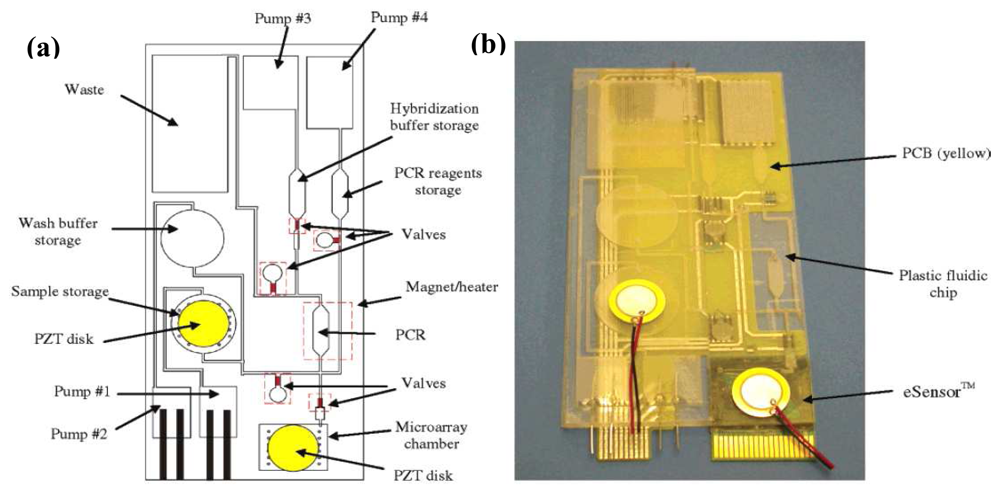
4. DNA Sensors
5. Conclusions
Acknowledgments
References
- Clark, L.C.; Lyons, C. Electrode Systems for Continuous Monitoring in Cardiovascular Surgery. Ann. NY Acad. Sci. 1962, 102, 29–45. [Google Scholar]
- D'Orazio, P. Biosensors in Clinical Chemistry. Clin. Chim. Acta 2003, 334, 41–69. [Google Scholar]
- Terry, L.A.; White, S.F.; Tigwell, L.J. The Application of Biosensors to Fresh Produce and the Wider Food Industry. J. Agr. Food Chem. 2005, 53, 1309–1316. [Google Scholar]
- Rodriguez-Mozaz, S.; Lopez de Alda, M.J.; Barcelo, D. Biosensors as Useful Tools for Environmental Analysis and Monitoring. Anal. Bioanal. Chem. 2006, 386, 1025–1041. [Google Scholar]
- Baeumner, A.J. Biosensors for Environmental Pollutants and Food Contaminants. Anal. Bioanal. Chem. 2003, 377, 434–445. [Google Scholar]
- Yu, D.; Blankert, B.; Vire, J.C.; Kauffmann, J.M. Biosensors in Drug Discovery and Drug Analysis. Anal. Lett. 2005, 38, 1687–1701. [Google Scholar]
- Gooding, J.J. Biosensor Technology for Detecting Biological Warfare Agents: Recent Progress and Future Trends. Anal. Chim. Acta 2006, 559, 137–151. [Google Scholar]
- Wilkins, E.; Atanasov, P. Glucose Monitoring: State of the Art and Future Possibilities. Med. Eng. Phys. 1996, 18, 273–288. [Google Scholar]
- Turner, A.P.F.; Chen, B.; Piletsky, S.A. In Vitro Diagnostics in Diabetes: Meeting the Challenge. Clin. Chem. 1999, 45, 1596–1601. [Google Scholar]
- Wang, J. Glucose Biosensors: 40 Years of Advances and Challenges. Electroanalysis 2001, 13, 983–988. [Google Scholar]
- Newman, J.D.; Turner, A.P.F. Home Blood Glucose Biosensors: A Commercial Perspective. Biosens. Bioelectron. 2005, 20, 2435–2453. [Google Scholar]
- Hall, E.A.H. Handbook of Biosensors and Biochips; Marks, R. S., Cullen, D. C., Karube, I., Lowe, C. R., Weetall, H. H., Eds.; Wiley: Chichester, 2007; Volume 2, Chapter 72; pp. 1111–1129. [Google Scholar]
- Updike, S.J.; Hicks, G.P. The Enzyme Electrode. Nature 1967, 214, 986–988. [Google Scholar]
- Guilbault, G.G.; Lubrano, G.J. An Enzyme Electrode for the Amperometric Determination of Glucose. Anal. Chim. Acta 1973, 64, 439–455. [Google Scholar]
- Schlapfer, P.; Mindt, W.; Racine, P.H. Electrochemical Measurement of Glucose Using Various Electron Acceptors. Clin. Chim. Acta 1974, 57, 283–289. [Google Scholar]
- Cass, A.E.G.; Davis, G.; Francis, G.D.; O., H.H.A.; Aston, W.J.; Higgins, I.J.; Plotkin, E.V.; Scott, L.D.L.; Turner, A.P.F. Ferrocene-Mediated Enzyme Electrode for Amperometric Determination of Glucose. Anal. Chem. 1984, 56, 667–671. [Google Scholar]
- Degani, Y.; Heller, A. Direct Electrical Communication between Chemically Modified Enzymes and Metal Electrodes. 1. Electron Transfer from Glucose Oxidase to Metal Electrodes via Electron Relays, Bound Covalently to the Enzyme. J. Phys. Chem. 1987, 91, 1285–1289. [Google Scholar]
- Chara, T.J.; Rajagopaian, R.; Heller, A. “Wired” Enzyme Electrodes for Amperometric Determination of Glucose or Lactate in the Presence of Interfering Substances. Anal. Chem. 1994, 66, 2451–2457. [Google Scholar]
- Zayats, M.; Katz, E.; Willner, I. Electrical Contacting of Glucose Oxidase by Surface-Reconstitution of the Apo-Protein on a Relay-Boronic Acid-FAD Cofactor Monolayer. J. Am. Chem. Soc. 2002, 124, 2120–2121. [Google Scholar]
- Raitman, O.A.; Katz, E.; Buckmann, A.F.; Willner, I. Integration of Polyaniline/Poly(acrylic acid) Films and Redox Enzymes on Electrode Supports: An in Situ Electrochemical/Surface Plasmon Resonance Study of the Bioelectrocatalyzed Oxidation of Glucose or Lactate in the Integrated Bioelectrocatalytic Systems. J. Am. Chem. Soc. 2002, 124, 6487–6496. [Google Scholar]
- Shichiri, M.; Kawamori, R.; Yamasaki, Y.; Hakui, N. Wearable Artificial Endocrine Pancreas with Needle-Type Glucose Sensor. Lancet 1982, 320, 1129–1131. [Google Scholar]
- Gough, D.A.; Lucisano, J.Y.; Tse, P.H.S. Two-Dimensional Enzyme Electrode Sensor for Glucose. Anal. Chem. 1985, 57, 2351–2357. [Google Scholar]
- Churchouse, S.J.; Battersby, C.M.; Mullen, W.H.; Vadgama, P.M. Needle Enzyme Electrodes for Biological Studies. Biosensors 1986, 2, 325–342. [Google Scholar]
- Turner, R.F.B.; Harrison, D.J.; Rajotte, R.V.; Baltes, H.P. A Biocompatible Enzyme Electrode for Continuous in Vivo Glucose Monitoring in Whole Blood. Sensor. Actuat. B-Chem. 1990, 1, 561–564. [Google Scholar]
- Bindra, D.S.; Zhang, Y.; Wilson, G.S.; Sternberg, R.; Thevenot, D.R.; Moatti, D.; Reach, G. Design and in Vitro Studies of a Needle-Type Glucose Sensor for Subcutaneous Monitoring. Anal. Chem. 1991, 63, 1692–1696. [Google Scholar]
- Shaw, G.W.; Claremont, D.J.; Pickup, J.C. In Vitro Testing of a Simply Constructed, Highly Stable Glucose Sensor Suitable for Implantation in Diabetic Patients. Biosens. Bioelectron. 1991, 6, 401–406. [Google Scholar]
- Moatti-Sirat, D.; Velho, G.; Reach, G. Evaluating in Vitro and in Vivo the Interference of Ascorbate and Acetaminophen on Glucose Detection by a Needle-Type Glucose Sensor. Biosens. Bioelectron. 1992, 7, 345–352. [Google Scholar]
- Kanapieniene, J.J.; Dedinaite, A.A.; Laurinavicius, V.S.A. Miniature Glucose Biosensor with Extended Linearity. Sensor. Actuat. B-Chem. 1992, 10, 37–40. [Google Scholar]
- Rhodes, R.K.; Shults, M.C.; Updike, S.J. Prediction of Pocket-Portable and Implantable Glucose Enzyme Electrode Performance from Combined Species Permeability and Digital Simulation Analysis. Anal. Chem. 1994, 66, 1520–1529. [Google Scholar]
- Abdel-Hamid, I.; Atanasov, P.; Wilkins, E. Development of a Needle Type Glucose Biosensor. Anal. Lett. 1994, 27, 1453–1473. [Google Scholar]
- Csoregi, E.; Schmidtke, D.W.; Heller, A. Design and Optimization of a Selective Subcutaneously Implantable Glucose Electrode Based on “Wired” Glucose Oxidase. Anal. Chem. 1995, 67, 1240–1244. [Google Scholar]
- Hashiguchi, Y.; Sakakida, M.; Nishida, K.; Uemura, T.; Kajiwara, K.; Shichiri, M. Development of Miniaturized Glucose Monitoring System by Combining a Needle-Type Glucose Sensor with Microdialysis Sampling Method. Diabetes Care 1994, 17, 387–396. [Google Scholar]
- Poscia, A.; Mascini, M.; Moscone, D.; Luzzana, M.; Caramenti, G.; Cremonesi, P.; Valgimigli, F.; Bongiovanni, C.; Varalli, M. A Microdialysis Technique for Continuous Subcutaneous Glucose Monitoring in Diabetic Patients (Part 1). Biosens. Bioelectron. 2003, 18, 891–898. [Google Scholar]
- Tamada, J.A.; Bohannon, N.J.V.; Potts, R.O. Measurement of Glucose in Diabetic Subjects Using Noninvasive Transdermal Extraction. Nat. Med. 1995, 1, 1198–1201. [Google Scholar]
- Tierney, M.J.; Tamada, J.A.; Potts, R.O.; Jovanovic, L.; Garg, S.; Team, C.R. Clinical Evaluation of the GluoWatch Biographer: A Continual, Non-Invasive Glucose Monitor for Patients with Diabetes. Biosens. Bioelectron. 2001, 16, 621–629. [Google Scholar]
- Pickup, J.C. Handbook of Biosensors and Biochips; Marks, R. S., Cullen, D. C., Karube, I., Lowe, C. R., Weetall, H. H., Eds.; Wiley: Chichester, 2007; Volume 2, Chapter 68; pp. 1069–1075. [Google Scholar]
- Zimmermann, S.; Fienbork, D.; Stoeber, B.; Flounders, A.W.; Liepmann, D. A Microneedle-Based Glucose Monitor: Fabricated on a Wafer-Level Using in-Deivce Enzyme Immobilization. Transducers '03, The 12th International Conference on Solid State Sensors, Actuators and Microsystems; 2003; pp. 99–102. [Google Scholar]
- Xiao, Y.; Patolsky, F.; Katz, E.; Hainfeld, J.F.; Willner, I. “Plugging into Enzymes”: Nanowiring of Redox Enzymes by a Gold Nanoparticle. Science 2003, 299, 1877–1881. [Google Scholar]
- Janata, J. An Immunoelectrode. J. Am. Chem. Soc. 1975, 97, 2914–2916. [Google Scholar]
- Tang, D.; Yuan, R.; Chai, Y.; Zhong, X.; Liu, Y.; Dai, J. Novel Potentiometric Immunosensor for the Detection of Diphtheria Antigen Based on Colloidal Gold and Polyvinyl Butyral as Matrixes. Biochem. Eng. J. 2004, 22, 43–49. [Google Scholar]
- Aizawa, M.; Morioka, A.; Suzuki, S.; Nagamura, Y. Enzyme Immunosensor: III. Amperometric Determination of Human Chorionic Gonadotropin by Membrane-Bound Antibody. Anal. Biochem. 1979, 94, 22–28. [Google Scholar]
- Meusel, M.; Renneberg, R.; Spener, F.; Schmitz, G. Development of a Heterogeneous Amperometric Immunosensor for the Determination of Apolipoprotein E in Serum. Biosens. Bioelectron. 1995, 10, 577–586. [Google Scholar]
- Kreuzer, M.P.; O'Sullivan, C.K.; Pravda, M.; Guilbault, G.G. Development of an Immunosensor for the Determination of Allergy Antibody (IgE) in Blood Samples. Anal. Chim. Acta 2001, 442, 45–53. [Google Scholar]
- O'Regan, T.; Pravda, M.; O'Sullivan, C.K.; Guilbault, G.G. Development of Biosensor Array for Rapid Detection of Cardiac Markers: Immunosensor for Detection of Free Cardiac Troponin I. Anal. Lett. 2003, 36, 1903–1920. [Google Scholar]
- Rao, V.K.; Rai, G.P.; Agarwal, G.S.; Suresh, S. Amperometric Immunosensor for Detection of Antibodies of Salmonella Typhi in Patient Serum. Anal. Chim. Acta 2005, 531, 173–177. [Google Scholar]
- Diaz-Gonzalez, M.; Gonzalez-Garcia, M.B.; Costa-Garcia, A. Detection of Pneumolysin in Human Urine Using an Immunosensor on Screen-Printed Carbon Electrodes. Sensor. Actuat. B-Chem. 2006, 113, 1005–1011. [Google Scholar]
- Akram, M.; Stuart, M.C.; Wong, D.K.Y. Signal Generation at an Electrochemical Immunosensor via the Direct Oxidation of an Electroactive Label. Electroanalysis 2006, 18, 237–246. [Google Scholar]
- Bataillard, P.; Gardies, F.; Jaffrezic-Renault, N.; Martelet, C. Direct Detection of Immunospecies by Capacitance Measurements. Anal. Chem. 1988, 60, 2374–2379. [Google Scholar]
- Mirsky, V.M.; Riepl, M.; Wolfbeis, O.S. Capacitive Monitoring of Protein Immobilization and Antigen-Antibody Reactions on Monomolecular Alkylthiol Films on Gold Electrodes. Biosens. Bioelectron. 1997, 12, 977–989. [Google Scholar]
- Taylor, R.F.; Marenchic, I.G.; Spencer, R.H. Antibody- and Receptor-Based Biosensors for Detection and Process Control. Anal. Chim. Acta 1991, 249, 67–70. [Google Scholar]
- Bardea, A.; Katz, E.; Willner, I. Probing Antigen–Antibody Interactions on Electrode Supports by the Biocatalyzed Precipitation of an Insoluble Product. Electroanalysis 2000, 12, 1097–1106. [Google Scholar]
- Schenck, J.F. Theory, Design and Biomedical Applications of Solid State Chemical Sensors; Cheung, P. W., Ed.; CRC Press: West Palm Beach, 1978; pp. 165–173. [Google Scholar]
- Besselink, G.A.J.; Schasfoort, R.B.M.; Bergveld, P. Modification of ISFETs with a Monolayer of Latex Beads for Specific Detection of Proteins. Biosens. Bioelectron. 2003, 18, 1109–1114. [Google Scholar]
- Gao, Q.; Ma, Y.; Cheng, Z.; Wang, W.; Yang, X. Flow Injection Electrochemical Enzyme Immunoassay Based on the Use of an Immunoelectrode Strip Integrate Immunosorbent Layer and a Screen-Printed Carbon Electrode. Anal. Chim. Acta 2003, 488, 61–70. [Google Scholar]
- Guan, J.G.; Miao, Y.Q.; Chen, J.R. Prussian Blue Modified Amperometric FIA Biosensor: One-Step Immunoassay for α-Fetoprotein. Biosens. Bioelectron. 2004, 19, 789–794. [Google Scholar]
- Duan, C.; Meyerhoff, M.E. Separation-Free Sandwich Enzyme Immunoassays Using Microporous Gold Electrodes and Self-Assembled Monolayer/Immobilized Capture Antibodies. Anal. Chem. 1994, 66, 1369–1377. [Google Scholar]
- Ivnitski, D.; Rishpon, J. A One-Step, Separation-Free Amperometric Enzyme Immunosensor. Biosens. Bioelectron. 1996, 11, 409–417. [Google Scholar]
- Darain, F.; Park, S.U.; Shim, Y.B. Disposable Amperometric Immunosensor System for Rabbit IgG Using a Conducting Polymer Modified Screen-Printed Electrode. Biosens. Bioelectron. 2003, 18, 773–780. [Google Scholar]
- Davies, R.J.; Eapen, S.S.; Carlisle, S.J. Handbook of Biosensors and Biochips; Marks, R. S., Cullen, D. C., Karube, I., Lowe, C. R., Weetall, H. H., Eds.; Wiley: Chichester, 2007; Volume 2, Chapter 74; pp. 1151–1165. [Google Scholar]
- Fernandez-Sanchez, C.; McNeil, C.J.; Rawson, K.; Nilsson, O. Disposable Noncompetitive Immunosensor for Free and Total Prostate-Specific Antigen Based on Capacitance Measurement. Anal. Chem. 2004, 76, 5649–5656. [Google Scholar]
- Fernandez-Sanchez, C.; Gallardo-Soto, A.M.; Rawson, K.; Nilsson, O.; McNeil, C.J. Quantitative Impedimetric Immunosensor for Free and Total Prostate Specific Antigen Based on a Lateral Flow Assay Format. Electrochem. Commun. 2004, 6, 138–143. [Google Scholar]
- Liu, G.; Lin, Y.Y.; Wang, J.; Wu, H.; Wai, C.M.; Lin, Y. Disposable Electrochemical Immunosensor Diagnosis Device Based on Nanoparticle Probe and Immunochromatographic Strip. Anal. Chem. 2007, 79, 7644–7653. [Google Scholar]
- Ikebukuro, K.; Kiyohara, C.; Sode, K. Novel Electrochemical Sensor System for Protein Using the Aptamers in Sandwich Manner. Biosens. Bioelectron. 2005, 20, 2168–2172. [Google Scholar]
- Bang, G.S.; GCho, S.; Kim, B.G. A Novel Electrochemical Detection Method for Aptamer Biosensors. Biosens. Bioelectron. 2005, 21, 863–870. [Google Scholar]
- Cai, H.; Lee, T.M.H.; Hsing, I.-M. Label-Free Protein Recognition Using an Aptamer-Based Impedance Measurement Assay. Sensor Actuat. B-Chem. 2006, 114, 433–437. [Google Scholar]
- Kawde, A.N.; Rodriguez, M.C.; Lee, T.M.H.; Wang, J. Label-Free Bioelectronic Detection of Aptamer-Protein Interactions. Electrochem. Commun. 2005, 7, 537–540. [Google Scholar]
- Xiao, Y.; Lubin, A.A.; Heeger, A.J.; Plaxco, K.W. Label-Free Electronic Detection of Thrombin in Blood Serum by Using an Aptamer-Based Sensor. Angew. Chem. Int. Edit. 2005, 44, 5456–5459. [Google Scholar]
- Radi, A.E.; Sanchez, J.L.A.; Baldrich, E.; O'Sullivan, C.K. Reagentless, Reusable, Ultrasensitive Electrochemical Molecular Beacon Aptasensor. J. Am. Chem. Soc. 2006, 128, 117–124. [Google Scholar]
- Xiao, Y.; Piorek, B.D.; Plaxco, K.W.; Heeger, A.J. A Reagentless Sign-On Architecture for Electronic, Aptamer-Based Sensors via Target-Induced Strand Displacement. J. Am. Chem. Soc. 2005, 127, 17990–17991. [Google Scholar]
- Rodriguez, M.C.; Kawde, A.N.; Wang, J. Aptamer Biosensor for Label-Free Impedance Spectroscopy Detection of Proteins Based on Recognition-Induced Switching of the Surface Charge. Chem. Commun. 2005, 4267–4269. [Google Scholar]
- Le Floch, F.; Ho, H.A.; Leclerc, M. Label-Free Electrochemical Detection of Protein Based on a Ferrocene-Bearing Cationic Polythiophene and Aptamer. Anal. Chem. 2006, 78, 4727–4731. [Google Scholar]
- Lai, R.Y.; Plaxco, K.W.; Heeger, A.J. Aptamer-Based Electrochemical Detection of Picomolar Platelet-Derived Growth Factor Directly in Blood Serum. Anal. Chem. 2007, 79, 229–233. [Google Scholar]
- Cheng, A.K.H.; Ge, B.; Yu, H.Z. Aptamer-Based Biosensors for Label-Free Voltammetric Detection of Lysozyme. Anal. Chem. 2007, 79, 5158–5164. [Google Scholar]
- Mir, M.; Katakis, I. Aptamers as Elements of Bioelectronic Devices. Mol. Biosyst. 2007, 3, 620–622. [Google Scholar]
- Lu, Y.; Li, X.; Zhang, L.; Yu, P.; Su, L.; Mao, L. Aptamer-Based Electrochemical Sensors with Aptamer-Complementary DNA Oligonucleotides as Probe. Anal. Chem. 2008, 80, 1883–1890. [Google Scholar]
- Zhang, Y.L.; Huang, Y.; Jiang, J.H.; Shen, G.L.; Yu, R.Q. Electrochemical Aptasensor Based on Proximity-Dependent Surface Hybridization Assay for Single-Step, Reusable, Sensitive Protein Detection. J. Am. Chem. Soc. 2007, 129, 15448–15449. [Google Scholar]
- Centi, S.; Tombelli, S.; Minunni, M.; Mascini, M. Aptamer-Based Detection of Plasma Proteins by an Electrochemical Assay Coupled to Magnetic Beads. Anal. Chem. 2007, 79, 1466–1473. [Google Scholar]
- Wang, J.; Ibanez, A.; Chatrathi, M.P.; Escarpa, A. Electrochemical Enzyme Immunoassays on Microchip Platforms. Anal. Chem. 2001, 73, 5323–5327. [Google Scholar]
- Zheng, G.; Patolsky, F.; Cui, Y.; Wang, W.U.; Lieber, C.M. Multiplexed Electrical Detection of Cancer Markers with Nanowire Sensor Arrays. Nat. Biotechnol. 2005, 23, 1294–1301. [Google Scholar]
- Chen, R.J.; Bangsaruntip, S.; Drouvalakis, K.A.; Kam, N.W.S.; Shim, M.; Li, Y.; Kim, W.; Utz, P.J.; Dai, H. Noncovalent Functionalization of Carbon Nanotubes for Highly Specific Electronic Biosensors. P. Natl. Acad. Sci. USA 2003, 100, 4984–4989. [Google Scholar]
- So, H.M.; Won, K.; Kim, Y.H.; Kim, B.K.; Ryu, B.H.; Na, P.S.; Kim, H.; Lee, J.O. Single-Walled Carbon Nanotube Biosensors Using Aptamers as Molecular Recognition Elements. J. Am. Chem. Soc. 2005, 127, 11906–11907. [Google Scholar]
- Maehashi, K.; Katsura, T.; Kerman, K.; Takamura, Y.; Matsumoto, K.; Tamiya, E. Label-Free Protein Biosensor Based on Aptamer-Modified Carbon Nanotube Field-Effect Transistors. Anal. Chem. 2007, 79, 782–787. [Google Scholar]
- Millan, K.M.; Mikkelsen, S.R. Sequence-Selective Biosensor for DNA Based on Electroactive Hybridization Indicators. Anal. Chem. 1993, 65, 2317–2323. [Google Scholar]
- Wang, J.; Rivas, G.; Cai, X.; Dontha, N.; Shiraishi, H.; Luo, D.; Valera, F.S. Sequence-Specific Electrochemical Biosensing of M. Tuberculosis DNA. Anal. Chim. Acta 1997, 337, 41–48. [Google Scholar]
- Hashimoto, K.; Ito, K.; Ishimori, Y. Sequence-Specific Gene Detection with a Gold Electrode Modified with DNA Probes and an Electrochemically Active Dye. Anal. Chem. 1994, 66, 3830–3833. [Google Scholar]
- Hashimoto, K.; Ito, K.; Ishimori, Y. Novel DNA Sensor for Electrochemical Gene Detection. Anal. Chim. Acta 1994, 286, 219–224. [Google Scholar]
- Marrazza, G.; Chianella, I.; Mascini, M. Disposable DNA Electrochemical Sensor for Hybridization Detection. Biosens. Bioelectron. 1999, 14, 43–51. [Google Scholar]
- Boon, E.M.; Ceres, D.M.; Drummond, T.G.; Hill, M.G.; Barton, J.K. Mutation Detection by Electrocatalysis at DNA-Modified Electrodes. Nat. Biotechnol. 2000, 18, 1096–1100. [Google Scholar]
- Erdem, A.; Kerman, K.; Meric, B.; Akarca, U.S.; Ozsoz, M. Novel Hybridization Indicator Methylene Blue for the Electrochemical Detection of Short DNA Sequences Related to the Hepatitis B Virus. Anal. Chim. Acta 2000, 422, 139–149. [Google Scholar]
- Wong, E.L.S.; Gooding, J.J. Electronic Detection of Target Nucleic Acids by a 2,6-Disulfonic Acid Anthraquinone Intercalator. Anal. Chem. 2003, 75, 3845–3852. [Google Scholar]
- Kelley, S.O.; Boon, E.M.; Barton, J.K.; Jackson, N.M.; Hill, M.G. Single-Base Mismatch Detection Based on Charge Transduction through DNA. Nucleic Acids Res. 1999, 27, 4830–4837. [Google Scholar]
- Yang, W.; Ozsoz, M.; Hibbert, D.B.; Gooding, J.J. Evidence for the Direct Interaction between Methylene Blue and Guanine Bases Using DNA-Modified Carbon Paste Electrodes. Electroanalysis 2002, 14, 1299–1302. [Google Scholar]
- Kerman, K.; Ozkan, D.; Kara, P.; Meric, B.; Gooding, J.J.; Ozsoz, M. Voltammetric Determination of DNA Hybridization Using Methylene Blue and Self-Assembled Alkanethiol Monolayer on Gold Electrodes. Anal. Chim. Acta 2002, 462, 39–47. [Google Scholar]
- Takenaka, S.; Yamashita, K.; Takagi, M.; Uto, Y.; Kondo, H. DNA Sensing on a DNA Probe-Modified Electrode Using Ferrocenylnaphthalene Diimide as the Electrochemically Active Ligand. Anal. Chem. 2000, 72, 1334–1341. [Google Scholar]
- Napier, M.E.; Loomis, C.R.; Sistare, M.F.; Kim, J.; Eckhardt, A.E.; Thorp, H.H. Probing Biomolecule Recognition with Electron Transfer: Electrochemical Sensors for DNA Hybridization. Bioconjugate Chem. 1997, 8, 906–913. [Google Scholar]
- Aoki, H.; Buhlmann, P.; Umezawa, Y. Electrochemical Detection of a One-Base Mismatch in an Oligonucleotide Using Ion-Channel Sensors with Self-Assembled PNA Monolayers. Electroanalysis 2000, 12, 1272–1276. [Google Scholar]
- Aoki, H.; Umezawa, Y. High Sensitive Ion-Channel Sensors for Detection of Oligonucleotides Using PNA Modified Gold Electrodes. Electroanalysis 2002, 14, 1405–1410. [Google Scholar]
- Patolsky, F.; Lichtenstein, A.; Willner, I. Electrochemical Transduction of Liposome-Amplified DNA Sensing. Angew. Chem. Int. Edit. 2000, 39, 940–943. [Google Scholar]
- Patolsky, F.; Katz, E.; Bardea, A.; Willner, I. Enzyme-Linked Amplified Electrochemical Sensing of Oligonucleotide–DNA Interactions by Means of the Precipitation of an Insoluble Product and Using Impedance Spectroscopy. Langmuir 1999, 15, 3703–3706. [Google Scholar]
- Campbell, C.N.; Gal, D.; Cristler, N.; Banditrat, C.; Heller, A. Enzyme-Amplified Amperometric Sandwich Test for RNA and DNA. Anal. Chem. 2002, 74, 158–162. [Google Scholar]
- Park, S.J.; Taton, A.; Mirkin, C.A. Array-Based Electrical Detection of DNA with Nanoparticle Probes. Science 2002, 295, 1503–1506. [Google Scholar]
- Ozsoz, M.; Erdem, A.; Kerman, K.; Ozkan, D.; Tugrul, B.; Topcuoglu, N.; Ekren, H.; Taylan, M. Electrochemical Genosensor Based on Colloidal Gold Nanoparticles for the Detection of Factor V Leiden Mutation Using Disposable Pencil Graphite Electrodes. Anal. Chem. 2003, 75, 2181–2187. [Google Scholar]
- Cai, H.; Wang, Y.; He, P.; Fang, Y. Electrochemical Detection of DNA Hybridization Based on Silver-Enhanced Gold Nanoparticle Label. Anal. Chim. Acta 2002, 469, 165–172. [Google Scholar]
- Lee, T.M.H.; Li, L.L.; Hsing, I.-M. Enhanced Electrochemical Detection of DNA Hybridization Based on Electrode-Surface Modification. Langmuir 2003, 19, 4338–4343. [Google Scholar]
- Lee, T.M.H.; Cai, H.; Hsing, I.-M. Gold Nanoparticle-Catalyzed Silver Electrodeposition on an Indium Tin Oxide Electrode and Its Application in DNA Hybridization Transduction. Electroanalysis 2004, 16, 1628–1631. [Google Scholar]
- Wang, J.; Liu, G.; Merkoci, A. Electrochemical Coding Technology for Simultaneous Detection of Multiple DNA Targets. J. Am. Chem. Soc. 2003, 125, 3214–3215. [Google Scholar]
- Liu, G.; Lee, T.M.H.; Wang, J. Nanocrystal-Based Bioelectronic Coding of Single Nucleotide Polymorphisms. J. Am. Chem. Soc. 2005, 127, 38–39. [Google Scholar]
- Umek, R.M.; Lin, S.W.; Vielmetter, J.; Terbrueggen, R.H.; Irvine, B.; Yu, C.J.; Blackburn, G.F.; Farkas, D.H.; Chen, Y.P. Electronic Detection of Nucleic Acids: A Versatile Platform for Molecular Diagnostics. J Mol. Diagn. 2001, 3, 74–84. [Google Scholar]
- Fan, C.; Plaxco, K.W.; Heeger, A.J. Electrochemical Interrogation of Conformational Changes as a Reagentless Method for the Sequence-Specific Detection of DNA. P. Natl. Acad. Sci. USA 2003, 100, 9134–9137. [Google Scholar]
- Immoos, C.E.; Lee, S.J.; Grinstaff, M.W. DNA-PEG-DNA Triblock Macromolecules for Reagentless DNA Detection. J. Am. Chem. Soc. 2004, 126, 10814–10815. [Google Scholar]
- Xiao, Y.; Lubin, A.A.; Baker, B.R.; Plaxco, K.W.; Heeger, A.J. Single-Step Electronic Detection of Femtomolar DNA by Target-Induced Strand Displacement in an Electrode-Bound Duplex. P. Natl. Acad. Sci. USA 2006, 103, 16677–16680. [Google Scholar]
- Wang, J.; Rivas, G.; Fernandes, J.R.; Paz, J.L.L.; Jiang, M.; Waymire, R. Indicator-Free Electrochemical DNA Hybridization Biosensor. Anal. Chim. Acta 1998, 375, 197–203. [Google Scholar]
- Ozkan, D.; Erdem, A.; Kara, P.; Kerman, K.; Meric, B.; Hassmann, J.; Ozsoz, M. Allele-Specific Genotype Detection of Factor V Leiden Mutation from Polymerase Chain Reaction Amplicons Based on Label-Free Electrochemical Genosensor. Anal. Chem. 2002, 74, 5931–5936. [Google Scholar]
- Lucarelli, F.; Marrazza, G.; Palchetti, I.; Cesaretti, S.; Mascini, M. Coupling of an Indicator-Free Electrochemical DNA Biosensor with Polymerase Chain Reaction for the Detection of DNA Sequences Related to the Apolipoprotein E. Anal. Chim. Acta 2002, 469, 93–99. [Google Scholar]
- Kerman, K.; Morita, Y.; Takamura, Y.; Tamiya, E. Label-Free Electrochemical Detection of DNA Hybridization on Gold Electrode. Electrochem. Commun. 2003, 5, 887–891. [Google Scholar]
- Guiducci, C.; Stagni, C.; Zuccheri, G.; Bogliolo, A.; Benini, L.; Samori, B.; Ricco, B. DNA Detection by Integrable Electronics. Biosens. Bioelectron. 2004, 19, 781–787. [Google Scholar]
- Fu, Y.; Yuan, R.; Xu, L.; Chai, Y.; Zhong, X.; Tang, D. Indicator-Free DNA Hybridization Detection via EIS Based on Self-Assembled Gold Nanoparticles and Bilayer Two-Dimensional 3-Mercaptopropyltrimethoxysilane onto a Gold Substrate. Biochem. Eng. J. 2005, 23, 37–44. [Google Scholar]
- Macanovic, A.; Marquette, C.; Polychronakos, C.; Lawrence, M.F. Impedance-Based Detection of DNA Sequences Using a Silicon Transducer with PNA as the Probe Layer. Nucleic Acids Res. 2004, 32, e20. [Google Scholar]
- Cai, W.; Peck, J.R.; van der Weide, D.W.; Hamers, R.J. Direct Electrical Detection of Hybridization at DNA-Modified Silicon Surfaces. Biosens. Bioelectron. 2004, 19, 1013–1019. [Google Scholar]
- Wang, J.; Jiang, M.; Fortes, A.; Mukherjee, B. New Label-Free DNA Recognition Based on Doping Nucleic-Acid Probes within Conducting Polymer Films. Anal. Chim. Acta 1999, 402, 7–12. [Google Scholar]
- Ramanaviciene, A.; Ramanavicius, A. Pulsed Amperometric Detection of DNA with an ssDNA/Polypyrrole-Modified Electrode. Anal. Bioanal. Chem. 2004, 379, 287–293. [Google Scholar]
- Komarova, E.; Aldissi, M.; Bogomolova, A. Direct Electrochemical Sensor for Fast Reagent-Free DNA Detection. Biosens. Bioelectron. 2005, 21, 182–189. [Google Scholar]
- Fritz, J.; Cooper, E.B.; Gaudet, S.; Sorger, P.K.; Manalis, S.R. Electronic Detection of DNA by Its Intrinsic Molecular Charge. P. Natl. Acad. Sci. USA 2002, 99, 14142–14146. [Google Scholar]
- Uslu, F.; Ingebrandt, S.; Mayer, D.; Bocker-Meffert, S.; Odenthal, M.; Offenhausser, A. Labelfree Fully Electronic Nucleic Acid Detection System Based on a Field-Effect Transistor Device. Biosens. Bioelectron. 2004, 19, 1723–1731. [Google Scholar]
- Kim, D.S.; Jeong, Y.T.; Park, H.J.; Shin, J.K.; Choi, P.; Lee, J.H.; Lim, G. An FET-Type Charge Sensor for Highly Sensitive Detection of DNA Sequence. Biosens. Bioelectron. 2004, 20, 69–74. [Google Scholar]
- Hou, C.S.J.; Milovic, N.; Godin, M.; Russo, P.R.; Chakrabarti, R.; Manalis, S.R. Label-Free Microelectronic PCR Quantification. Anal. Chem. 2006, 78, 2526–2531. [Google Scholar]
- Hahm, J.I.; Lieber, C.M. Direct Ultrasensitive Electrical Detection of DNA and DNA Sequence Variations Using Nanowire Nanosensors. Nano Lett. 2004, 4, 51–54. [Google Scholar]
- Gao, Z.; Agarwal, A.; Trigg, A.D.; Singh, N.; Fang, C.; Tung, C.H.; Fan, Y.; Buddharaju, K.D.; Kong, J. Silicon Nanowire Arrays for Label-Free Detection of DNA. Anal. Chem. 2007, 79, 3291–3297. [Google Scholar]
- Liu, R.H.; Yang, J.; Lenigk, R.; Bonanno, J.; Grodzinski, P. Self-Contained, Fully Integrated Biochip for Sample Preparation, Polymerase Chain Reaction Amplification, and DNA Microarray Detection. Anal. Chem. 2004, 76, 1824–1831. [Google Scholar]
- Yeung, S.S.W.; Lee, T.M.H.; Hsing, I.-M. Electrochemistry-Based Real-Time PCR on a Microchip. Anal. Chem. 2008, 80, 363–368. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbott FreeStyle Lite | Bayer Contour | LifeScan OneTouch Ultra2 | Roche Accu-Chek Aviva | |
|---|---|---|---|---|
| Sample Size | 0.3 μL | 0.6 μL | 1 μL | 0.6 μL |
| Test Range | 1.1 – 27.8 mM | 0.6 – 33.3 mM | 1.1 – 33.3 mM | 0.6 – 33.3 mM |
| Test Time | 5 s | 5 s | 5 s | 5 s |
| Alternative Site Testing | Hand, forearm, upper arm, thigh, or calf | Palm or forearm | Palm or forearm | Palm, forearm, upper arm, thigh, or calf |
| Memory | 400 results | 480 results | 500 results | 500 results |
| Special Feature | No coding required | No coding required, 7, 14, and 30-day averages | Link after meal results with food and portion choices | 7, 14, and 30-day averages |
| ApexBio The Edge | Arkray Lactate Pro | EKF Diagnostic Lactate Scout | Nova Biomedical Lactate Plus | |
|---|---|---|---|---|
| Sample Size | 3 μL | 5 μL | 0.5 μL | 0.7 μL |
| Test Range | 1.1 – 22.2 mM | 0.8 – 23.3 mM | 0.5 – 25.0 mM | 0.3 – 25.0 mM |
| Test Time | 45 s | 60 s | 15 s | 13 s |
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, T.M.-H. Over-the-Counter Biosensors: Past, Present, and Future. Sensors 2008, 8, 5535-5559. https://doi.org/10.3390/s8095535
Lee TM-H. Over-the-Counter Biosensors: Past, Present, and Future. Sensors. 2008; 8(9):5535-5559. https://doi.org/10.3390/s8095535
Chicago/Turabian StyleLee, Thomas Ming-Hung. 2008. "Over-the-Counter Biosensors: Past, Present, and Future" Sensors 8, no. 9: 5535-5559. https://doi.org/10.3390/s8095535
APA StyleLee, T. M. -H. (2008). Over-the-Counter Biosensors: Past, Present, and Future. Sensors, 8(9), 5535-5559. https://doi.org/10.3390/s8095535