Photoreactions and Structural Changes of Anabaena Sensory Rhodopsin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
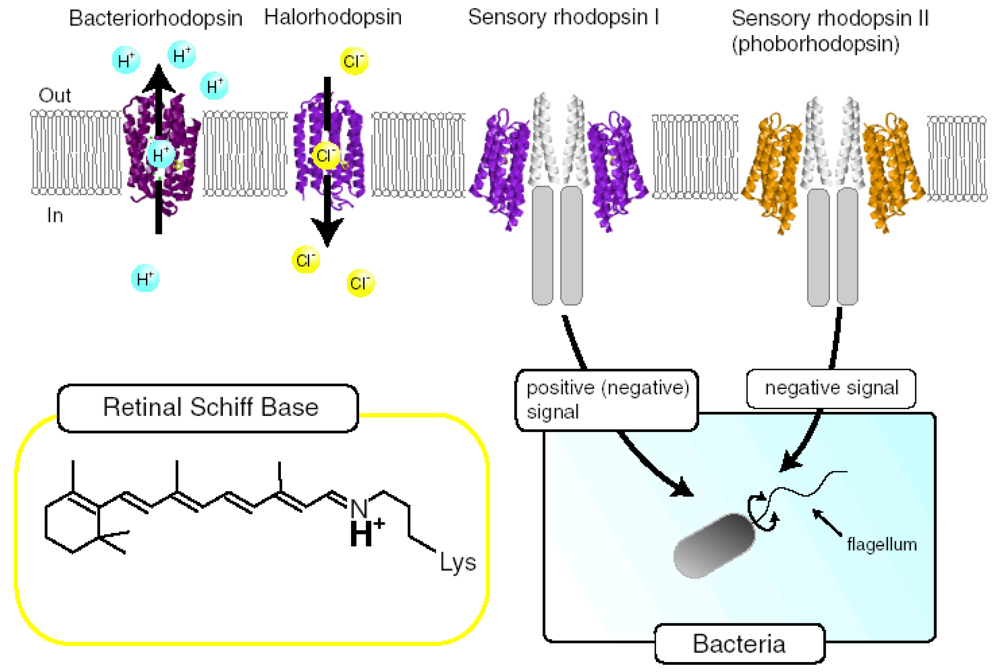
:1. Introduction
2. FTIR Spectroscopy of the All-trans Form of Anabaena Sensory Rhodopsin at 77 K: Hydrogen Bond of a Water between the Schiff Base and Asp75
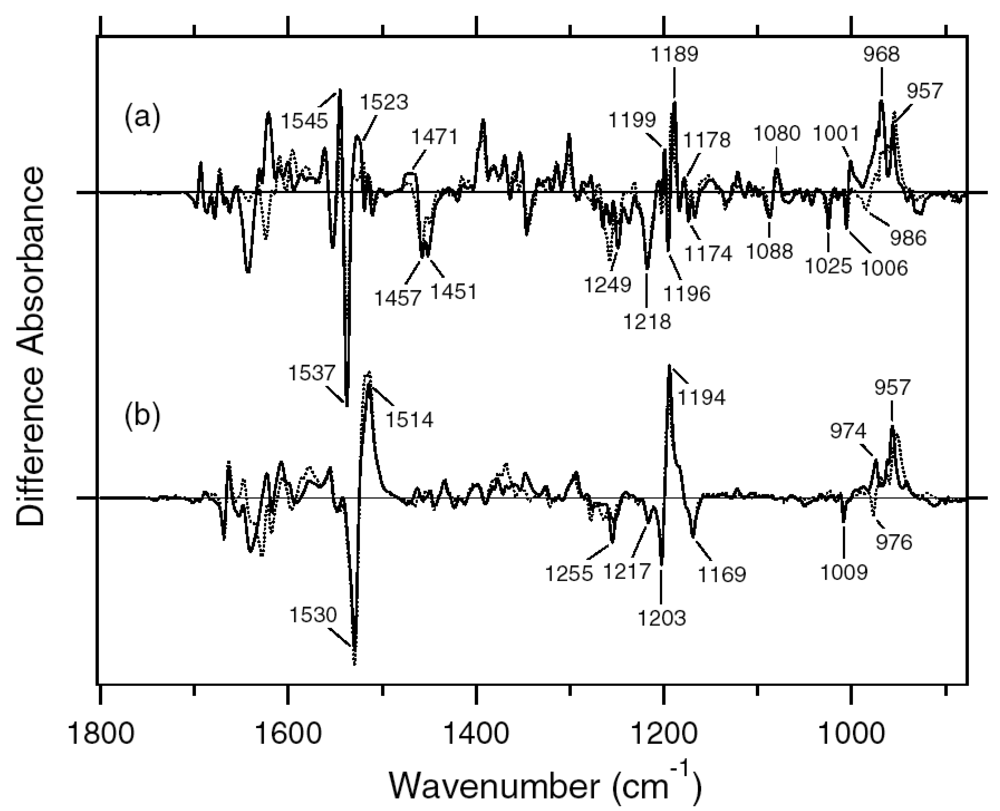
2.1. Comparison of the Difference Infrared Spectra Obtained by the Photoreactions of Anabaena Sensory Rhodopsin (ASR) at 77 K with Those of Bacteriorhodopsin (BR)
2.2. Comparison of the Vibrational Bands of the Retinal Chromophore between ASR and BR
2.3. Comparison of the C=O Stretching Vibrations of Carboxylate, Carbonyl, and Amide Groups of the Protein Moiety between ASR and BR
2.4. Comparison of the X-D Stretching Vibrations between ASR and BR
2.5. S-H Stretching Vibrations of the Cysteine Residues
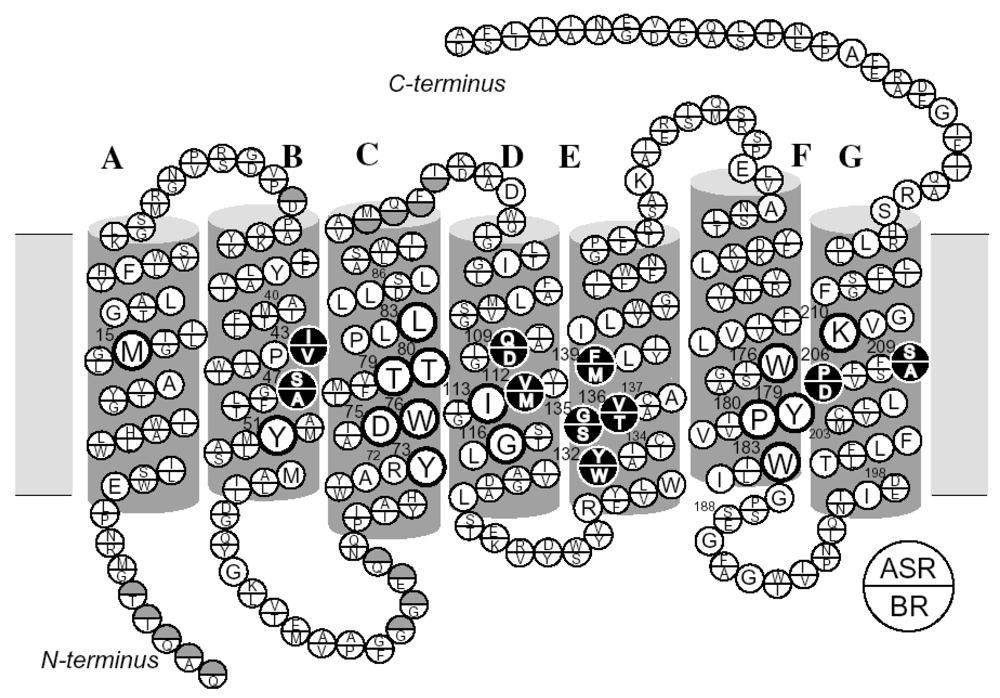
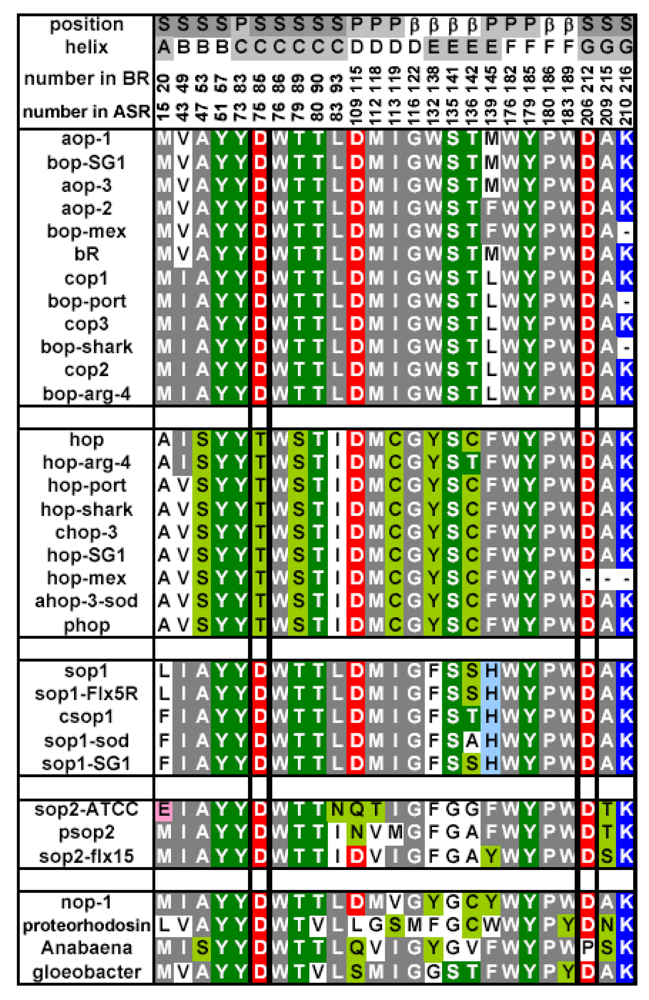
2.6. Unique Structure of the All-trans Form of ASR
3. FTIR Study of the Photoisomerization Processes in the 13-cis and All-trans Forms of Anabaena Sensory Rhodopsin at 77 K
3.1. Comparison of the Difference Infrared Spectra of the Photoreactions of 13C-ASR and AT-ASR at 77 K in the 1,770–870 cm-1 Region
3.2. S-H Stretching Vibrations of Cysteine Residues
3.3. Assignment of the N-D Stretching Vibrations in 13C-ASR and AT-ASR
3.4. O-D Stretching Vibrations of Water in 13C-ASR and AT-ASR
3.5. Unique Structure of the 13-cis Form of ASR
3.6. Unphotolyzed State of 13C-ASR
3.7. Photoisomerization Process of 13C-ASR in Comparison with that of AT-ASR
4. Photochromism of Anabaena Sensory Rhodopsin
4.1. Photoconversion of ASRAT (1) Photoreaction at 170 K
4.2. Photoconversion of ASRAT (2) Thermal Relaxation by warming the Sample from 170 K to 277 K
4.3. Photoconversion of ASR13C (1) Relative Photoconversion Yields of ASRAT and ASR13C at 277 K
4.4. Photoconversion of ASR13C (2) Relative Photoisomerization Quantum Yields of ASRAT and ASR13C at 130 K
4.5. Functional Optimization of Photoconversions in Rhodopsins
5. FTIR Study of the L Intermediate of Anabaena Sensory Rhodopsin: Structural Changes in the Cytoplasmic Region
5.1. Comparison of the Difference Infrared Spectra of the L Intermediate of Full-length ASR and Truncated ASR in the 1,800–800 cm-1 Region
5.2. Comparison of the Difference Infrared Spectra of the L Intermediate of ASR and BR in the 1,800–800 cm-1 Region
5.3. Comparison of the Difference Infrared Spectra of the L Intermediate in Protonated Carboxylic Acid (1,800–1,700 cm-1) Region
5.4. Comparison of the Difference Infrared Spectra of the L Intermediate in Water O-D Stretching Frequency (2,750–2,500 cm-1) Region
5.5. The Structure of ASRL
5.6. Hydrogen-Bonding Structures in the Cytoplasmic Domain of ASR and ASRL
5.7. Characteristic Features of Photoreaction in ASR
6. Experimental Section
ASR Sample Preparation
6.1. FTIR Spectroscopy
6.2. Accumulation of AT-ASRK
6.3. Accumulation of 13C-ASRK
6.4. Accumulation of ASRL
6.5. UV-Visible Spectroscopy
6.6. HPLC Analysis
7. Conclusions and Perspectives
Acknowledgments
References and Notes
- Lanyi, J.K. Understanding structure and function in the light-driven proton pump bacteriorhodopsin. J. Struct. Biol. 1998, 124, 164–178. [Google Scholar]
- Essen, L.O. Halorhodopsin: light-driven ion pumping made simple. Curr. Opsin. Struct. Biol. 2002, 12, 516–522. [Google Scholar]
- Kamo, N.; Shimono, K.; Iwamoto, M.; Sudo, Y. Photochemistry and photoinduced proton-transfer by pharaonis phoborhodopsin. Biochemistry (Mosc) 2001, 66, 1277–1282. [Google Scholar]
- Sasaki, J.; Spudich, J.L. Proton transport by sensory rhodopsins and its modulation by transducer-binding. Biochim. Biophys. Acta 2000, 1460, 230–239. [Google Scholar]
- Haupts, U.; Tittor, J.; Oesterhelt, D. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 367–399. [Google Scholar]
- Lanyi, J.K. Molecular Mechanism of Ion Transport in Bacteriorhodopsin: Insights from Crystallographic, Spectroscopic, Kinetic, and Mutational Studies. J. Phys. Chem. B 2000, 104, 11441–11448. [Google Scholar]
- Luecke, H.; Schobert, B.; Richter, H.T.; Cartailler, J.P.; Lanyi, J.K. Structure of Bacteriorhodopsin at 1.55 Å Resolution. J. Mol. Biol. 1999, 291, 899–911. [Google Scholar]
- Bieszke, J.A.; Braun, E.L.; Bean, L.E.; Kang, S.; Natvig, D.O.; Borkovich, K.A. The nop-1 gene of Neurospora crassa encodes a seven transmembrane helix retinal-binding protein homologous to archaeal rhodopsins. Proc. Natl. Acad. Sci. USA 1999, 96, 8034–8039. [Google Scholar]
- Sineshchekov, O.A.; Jung, K.H.; Spudich, J.L. Two rhodopsins mediate phototaxis to low- and high-intensity light in Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 2002, 99, 8689–8694. [Google Scholar]
- Nagel, G.; Ollig, D.; Fuhrmann, M.; Kateriya, S.; Musti, A.M.; Bamberg, E.; Hegemann, P. Channelrhodopsin-1: a light-gated proton channel in green algae. Science 2002, 296, 2395–2398. [Google Scholar]
- Okamoto, O.K.; Hastings, J.W. Novel dinoflagellate clock-related genes identified through microarray analysis. J. Phycol. 2003, 39, 519–526. [Google Scholar]
- Jung, K.H.; Spudich, J.L. Microbial rhodopsins: Transport and sensory proteins throughout the three domains of life. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd Ed.; Horspool, W.M., Lenci, F., Eds.; CRC: Boca Raton, FL, USA, 2004; pp. 124:1–124:11. [Google Scholar]
- de la Torre, J.R.; Christianson, L.M.; Beja, O.; Suzuki, M.T.; Karl, D.M.; Heidelberg, J.; DeLong, E.F. Proteorhodopsin genes are distributed among divergent marine bacterial taxa. Proc. Natl. Acad. Sci. USA 2003, 100, 12830–12835. [Google Scholar]
- Beja, O.; Aravind, L.; Koonin, E.V.; Suzuki, M.T.; Hadd, A.; Nguyen, L.P.; Jovanovich, S.B.; Gates, C.M.; Feldman, R.A.; Spudich, J.L.; Spudich, E.N.; DeLong, E.F. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 2000, 289, 1902–1906. [Google Scholar]
- Jung, K.H.; Trivedi, V.D.; Spudich, J.L. Demonstration of a sensory rhodopsin in eubacteria. Mol. Microbiol. 2003, 47, 1513–1522. [Google Scholar]
- Vogeley, L.; Sineshchekov, O.A.; Trivedi, V.D.; Sasaki, J.; Spudich, J.L.; Luecke, H. Anabaena sensory rhodopsin: a photochromic color sensor at 2.0 A. Science 2004, 306, 1390–1393. [Google Scholar]
- Vogeley, L.; Trivedi, V.D.; Sineshchekov, O.A.; Spudich, E.N.; Spudicu, J.L.; Luecke, H. Crystal structure of the Anabaena sensory rhodopsin transducer. J. Mol. Biol. 2007, 367, 741–751. [Google Scholar]
- Grossman, A.R.; Bhaya, D.; He, Q. Tracking the light environment by cyanobacteria and the dynamic nature of light harvesting. J. Biol. Chem. 2001, 276, 11449–11452. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–23. [Google Scholar]
- Furutani, Y.; Kawanabe, A.; Jung, K.H.; Kandori, H. FTIR spectroscopy of the all-trans form of Anabaena sensory rhodopsin at 77K: hydrogen bond of a water between the schiff base and Asp 75. Biochemistry 2005, 44, 12287–12296. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–80. [Google Scholar]
- Kandori, H. Role of internal water molecules in Bacteriorhodopsin. Biochem. Biophys. Acta 2000, 1460, 177–191. [Google Scholar]
- Kandori, H. Hydration switch model for the proton transfer in the Schiff base region of Bacteriorhodopsin. Biochem. Biophys. Acta 2004, 1658, 72–79. [Google Scholar]
- Iwamoto, M.; Shimono, K.; Sumi, M.; Kamo, N. Positioning proton-donating residues to the Schiff-base accelerates the M-decay of pharaonis phoborhodopsin expressed i. Escherichia coli. Biophys. Chem. 1999, 79, 187–92. [Google Scholar]
- Shibata, M.; Tanimoto, T.; Kandori, H. Water molecules in the schiff base region of bacteriorhodopsin. J. Am. Chem. Soc. 2003, 125, 13312–13313. [Google Scholar]
- Tanimoto, T.; Furutani, Y.; Kandori, H. Structural changes of water in the Schiff base region of bacteriorhodopsin: proposal of a hydration switch model. Biochemistry 2003, 42, 2300–2306. [Google Scholar]
- Furutani, Y.; Shibata, M.; Kandori, H. Strongly hydrogen-bonded water molecules in the Schiff base region of rhodopsins. Photochem. Photobiol. Sci. 2005, 9, 661–666. [Google Scholar]
- Kandori, H.; Kinoshita, N.; Shichida, Y.; Maeda, A. Protein Structural Changes in Bacteriorhodopsin upon Photoisomerization As Revealed by Polarized FTIR Spectroscopy. J. Phys. Chem. B 1998, 102, 7899–7905. [Google Scholar]
- Aton, B.; Doukas, A.G.; Callender, R.H.; Becher, B.; Ebrey, T.G. Resonance Raman studies of the purple membrane. Biochemistry 1997, 16, 2995–2999. [Google Scholar]
- Rothschild, K.J.; Bousche, O.; Braiman, M.S.; Hasselbacher, C.A.; Spudich, J.L. Fourier transform infrared study of the halorhodopsin chloride pump. Biochemistry 1988, 27, 2420–2424. [Google Scholar]
- Smith, S.O.; Braiman, M.S.; Myers, A.B.; Pardoen, J.A.; Courtin, J.M.L.; Winkel, C.; Lugtenburg, J.; Mathies, R.A. Vibrational Analysis of the all-trans-Retinal Chromophore in Light-Adapted Bacteriorhodopsin. J. Am. Chem. Soc. 1987, 109, 3108–3125. [Google Scholar]
- Braiman, M.; Mathies, R. Resonance Raman spectra of bacteriorhodopsin's primary photoproduct: evidence for a distorted 13-cis retinal chromophore. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 403–407. [Google Scholar]
- Roepe, P.D.; Ahl, P.L.; Herzfeld, J.; Lugtenburg, J.; Rothschild, K.J. Tyrosine Protonation Changes in Bacteriorhodopsin. J. Biol. Chem. 1988, 263, 5110–5117. [Google Scholar]
- Maeda, A.; Sasaki, J.; Pfefferle, J.M.; Shichida, Y.; Yoshizawa, T. Fourier transform infrared spectral studies on the Schiff base mode of all-trans bacteriorhodopsin and its photointermediates, K and L. Photochem. Photobiol. 1991, 54, 911–921. [Google Scholar]
- Maeda, A.; Balashov, S.P.; Lugtenburg, J.; Verhoeven, M.A.; Herzfeld, J.; Belenky, M.; Gennis, R.B.; Tomson, F.L.; Ebrey, T.G. Interaction of Internal Water Molecules with the Schiff Base in the L Intermediate of the Bacteriorhodopsin Photocycle. Biochemistry 2002, 41, 3803–3809. [Google Scholar]
- Kandori, H.; Shimono, K.; Sudo, Y.; Iwamoto, M.; Shichida, Y.; Kamo, N. Structural changes of pharaonis phoborhodopsin upon photoisomerization of the retinal chromophore: infrared spectral comparison with bacteriorhodopsin. Biochemistry 2001, 40, 9238–46. [Google Scholar]
- Furutani, Y.; Bezerra, A.G., Jr; Waschuk, S.; Sumii, M.; Brown, L.S.; Kandori, H. FTIR Spectroscopy of the K Photointermediate of Neurospora Rhodopsin: Structural Changes of the Retinal, Protein, and Water Molecules after Photoisomerization. Biochemistry 2004, 43, 9636–9646. [Google Scholar]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: San Diego, CA, USA, 1991. [Google Scholar]
- Rodman-Gilson, H.S.; Honig, B.; Croteau, A.; Zarrilli, G.; Nakanishi, K. Analysis of the factors that influence the C=N stretching frequency of polyene Schiff bases. Implications for bacteriorhodopsin and rhodopsin. Biophys. J. 1988, 53, 261–269. [Google Scholar]
- Baasov, T.; Friedman, N.; Sheves, M. Factors Affecting the C=N Stretching in Protonated Retinal Schiff Base: A Model Study for Bacteriorhodspsin and Visual Pigments? Biochemistry 1987, 26, 3210–3217. [Google Scholar]
- Kandori, H.; Belenky, M.; Herzfeld, J. Vibrational frequency and dipolar orientation of the protonated Schiff base in bacteriorhodopsin before and after photoisomerization. Biochemistry 2002, 41, 6026–6031. [Google Scholar]
- Braiman, M.S.; Mogi, T.; Marti, T.; Stern, L.J.; Khorana, H.G.; Rothschild, K.J. Vibrational spectroscopy of Bacteriorhodopsin mutants: Light-driven proton transport involves protonation changes of aspartic acid residues 85, 96, and 212. Biochemistry 1988, 27, 8516–8520. [Google Scholar]
- Kandori, H.; Shimono, K.; Shichida, Y.; Kamo, N. Interaction of Asn105 with the Retinal Chromophore during Photoisomerization of pharaonis Phoborhodopsin. Biochemistry 2002, 41, 4554–4559. [Google Scholar]
- Krimm, S.; Dwivedi, A.M. Infrared spectrum of the purple membrane: clue to a proton conduction mechanism? Science 1982, 216, 407–408. [Google Scholar]
- Yamazaki, Y.; Tuzi, S.; Saito, H.; Kandori, H.; Needleman, R.; Lanyi, J.K.; Maeda, A. Hydrogen Bonds of Water and C=O Groups Coordinate Long-Range Structural Changes in the L Photointermediate of Bacteriorhodopsin. Biochemistry 1996, 35, 4063–4068. [Google Scholar]
- Kandori, H.; Shichida, Y. Direct Observation of the Bridged Water Stretching Vibrations Inside a Protein. J. Am. Chem. Soc. 2000, 122, 11745–11746. [Google Scholar]
- Hayashi, S.; Tajkhorshid, E.; Kandori, H.; Schulten, K. Role of Hydrogen-Bond Network in Energy Storage of Bacteriorhodopsin's Light-Driven Proton Pump Revealed by ab Initio Normal-Mode Analysis. J. Am. Chem. Soc. 2004, 126, 10516–10517. [Google Scholar]
- Kandori, H.; Yamazaki, Y.; Shichida, Y.; Raap, J.; Lugtenburg, J.; Belenky, M.; Herzfeld, J. Tight Asp-85–Thr-89 association during the pump switch of Bacteriorhodopsin. Proc. Natl. Acad. Sci. USA 2001, 98, 1571–1576. [Google Scholar]
- Kandori, H.; Kinoshita, N.; Yamazaki, Y.; Maeda, A.; Shichida, Y.; Needleman, R.; Lanyi, J.K.; Bizounok, M.; Herzfeld, J.; Raap, J.; Lugtenburg, J. Structural Change of Threonine 89 upon Photoisomerization in Bacteriorhodopsin As Revealed by Polarized FTIR Spectroscopy. Biochemistry 1999, 38, 9676–9683. [Google Scholar]
- Takeuchi, H.; Harada, I. Ultraviolet resonance Raman spectroscopy of X-proline bonds: A new marker band of hydrogen bonding at the imide C=O site. J. Raman Spectrosc. 1990, 21, 509–515. [Google Scholar]
- Rothschild, K.J.; He, Y.W.; Gray, D.; Roepe, P.D.; Pelletier, S.L.; Brown, R.S.; Herzfeld, J. Fourier transform infrared evidence for proline structural changes during the bacteriorhodopsin photocycle. Proc. Natl. Acad. Sci. USA 1989, 86, 9832–9835. [Google Scholar]
- Royant, A.; Nollert, P.; Edman, K.; Neutze, R.; Landau, E.M.; Pebay-Peyroula, E.; Navarro, J. X-ray structure of sensory rhodopsin II at 2.1-Å resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 10131–10136. [Google Scholar]
- Luecke, H.; Schobert, B.; Lanyi, J.K.; Spudich, E.N.; Spudich, J.L. Crystal Structure of Sensory Rhodopsin II at 2.4 Angstroms: Insights into Color Tuning and Transducer Interaction. Science 2001, 293, 1499–1503. [Google Scholar]
- Shibata, M.; Kandori, H. FTIR Studies of Internal Water Molecules in the Schiff Base Region of Bacteriorhodopsin. Biochemistry 2005, 44, 7406–7413. [Google Scholar]
- Shibata, M.; Muneda, N.; Ihara, K.; Sasaki, T.; Demura, M.; Kandori, H. Internal water molecules of light-driven chloride pump proteins. Chem. Phys. Lett. 2004, 392, 330–333. [Google Scholar]
- Furutani, Y.; Shichida, Y.; Kandori, H. Structural Changes of Water Molecules during the Photoactivation Processes in Bovine Rhodopsin. Biochemistry 2003, 42, 9619–9625. [Google Scholar]
- Shineshchekov, O.A.; Trivedi, V.D.; Sasaki, J.; Spudich, J.L. Photochromicity of Anabaena sensory rhodopsin, an atypical microbial recepter with a cis-retinal light-adapted form. J. Biol. Chem. 2005, 280, 14663–14668. [Google Scholar]
- Pettei, J.M.; Yudd, P.A.; Nakanishi, K.; Henselman, R.; Stoeckenius, W. Identification of retinal isomers isolated from bacteriorhodopsin. Biochemistry 1977, 16, 1955–1959. [Google Scholar]
- Kawanabe, A.; Furutani, Y.; Jung, K.H.; Kandori, H. FTIR study of the photoisomerization processes in the 13-cis and all-trans forms of Anabaena sensory rhodopsin at 77 K. Biochemistry 2006, 45, 4362–4370. [Google Scholar]
- Mathies, R.A.; Smith, S.O.; Pollard, W.T. Biological applications of Raman spectroscopy: Resonance Raman spectra of polyenes and aromatics; Spiro, T.G., Ed.; John Wiley and Sons: New York, NY, USA, 1987; pp. 59–108. [Google Scholar]
- Eisenberg, D.; Kauzmann, W. The Structure and Properties of Water; Oxford University: London, UK, 1969. [Google Scholar]
- Birge, R.R. Nature of the primary photochemical events in rhodopsin and bacteriorhodopsin. Biochim. Biophys. Acta 1990, 1016, 293–327. [Google Scholar]
- Spudich, J.L.; Yang, C.S.; Jung, K.H.; Spudich, E.N. Retinylidene Proteins: Structures and Functions from Archaea to Humans. Annu. Rev. Cell. Dev. Biol. 2000, 16, 365–392. [Google Scholar]
- Kandori, H. Retinal Binding Proteins: From cis-trans Isomerization in Biochemistry; Wiley-VCH: Freiburg, Germany, 2006; Volume Chapter 4. [Google Scholar]
- Kochendoerfer, G.G.; Lin, S.W.; Sakmar, T.P.; Mathies, R. How color visual pigments are tuned. Trends. Biochem. Sci. 1999, 24, 300–305. [Google Scholar]
- Kandori, H.; Shichida, Y.; Yoshizawa, T. Photoisomerization in Rhodopsin. Biochemistry (Moscow) 2001, 66, 1197–1209. [Google Scholar]
- Scherrer, P.; Mathew, M.K.; Sperling, W.; Stoecknius, W. Retinal Isomer Ratio in Dark-Adapted Purple Membrane and Bacteriorhodopsin Monomers. Biochemistry 1989, 28, 829–834. [Google Scholar]
- Waschuk, S.K.; Bezerra, A.G., Jr; Shi, L.; Brown, L.S. Leptosphaeria rhodopsin: Bacteriorhodopsin-like proton pump from a eukaryote. Proc. Natl. Acad. Sci. USA 2005, 102, 6879–6883. [Google Scholar]
- Imamoto, Y.; Shichida, Y.; Hirayama, J.; Tomioka, H.; Kamo, N.; Yoshizawa, T. Chromophore Configuration of pharaonis Phoborhodopsin and Its Isomerization on Photon Absorption. Biochemistry 1992, 31, 2523–2528. [Google Scholar]
- Tsuda, M.; Nelson, B.; Chang, C.H.; Govindjee, R.; Ebrey, T.G. Characterization of the chromophore of the third rhodopsin-like pigment of Halobacterium halobium and its photoproduct. Biophys. J. 1985, 47, 721–724. [Google Scholar]
- Kawanabe, A.; Furutani, Y.; Jung, K.H.; Kandori, H. Photochromism of Anabaena sensory rhodopsin. J. Am. Chem. Soc. 2007, 129, 8644–8649. [Google Scholar]
- Sudo, Y.; Spudich, J.L. Three strategically placed hydrogen-bonding residues convert a proton pump into a sensory receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 16129–16134. [Google Scholar]
- Bergo, V.B.; Ntefidou, M.; Trivedi, V.D.; Amsden, J.J.; Kralj, J.M.; Rothschild, K.J.; Spudich, J.L. Conformational changes in the photocycle of Anabaena sensory rhodopsin: absence of the Schiff base counterion protonation signal. J. Biol. Chem. 2006, 281, 15208–15214. [Google Scholar]
- Shi, L.; Yoon, S.R.; Bezerra, A.G., Jr; Jung, K.H.; Brown, L.S. Cytoplasmic shuttling of protons in Anabaena sensory rhodopsin: implications for signaling mechanism. J. Mol. Biol. 2006, 358, 686–700. [Google Scholar]
- Shibata, M.; Yoshitsugu, M.; Mizuide, N.; Ihara, K.; Kandori, H. Halide binding by the D212N mutant of Bacteriorhodopsin affects hydrogen bonding of water in the active site. Biochemistry 2007, 46, 7525–7535. [Google Scholar]
- Sineshchekov, O.A.; Spudich, E.N.; Trivedi, V.D.; Spudich, J.L. Role of the cytoplasmic domain in Anabaena sensory rhodopsin photocycling: vectoriality of Schiff base deprotonation. Biophys. J. 2006, 91, 4519–4527. [Google Scholar]
- Kawanabe, A.; Furutani, Y.; Jung, K.H.; Kandori, H. FTIR study of the L-intermediate of Anabaena sensory rhodopsin: Structural changes in the cytoplasmic region. Biochemistry 2008, 47, 10033–10040. [Google Scholar]
- Gerwert, K.; Siebert, F. Evidence for light-induced 13-cis, 14-s-cis isomerization in bacteriorhodopsin obtained by FTIR difference spectroscopy using isotopically labelled retinals. EMBO J. 1986, 5, 805–811. [Google Scholar]
- Lorenz-Fonfria, V.A.; Furutani, Y.; Kandori, H. Active internal waters in the bacteriorhodopsin photocycle. A comparative study of the L and M intermediates at room and cryogenic temperatures by infrared spectroscopy. Biochemistry 2008, 47, 4071–4081. [Google Scholar]
- Litvin, F.F.; Balashov, S.P.; Sineshchekov, V.A. The investigation of the primary photochemical conversions of bacteriorhodopsin in purple membranes and cells of Halobacterium halobium by the low temperature spectrophotometry method. Bioorg. Khim. 1975, 1, 1767–1777. [Google Scholar]
- Dioumaev, A.K.; Lanyi, J.K. Bacteriorhodopsin photocycle at cryogenic temperatures reveals distributed barriers of conformational substates. Proc. Natl. Acad. Sci. USA 2007, 104, 9621–9626. [Google Scholar]
- Rammelsberg, R.; Huhn, G.; Lubben, M.; Gerwert, K. Bacteriorhodopsin's intramolecular proton-release pathway consists of a hydrogen-bonded network. Biochemistry 1998, 37, 5001–5009. [Google Scholar]
- Garczarek, F.; Brown, L.S.; Lanyi, J.K.; Gerwert, K. Proton binding within a membrane protein by a protonated water cluster. Proc. Natl. Acad. Sci. USA 2004, 102, 3633–3638. [Google Scholar]
- Choi, A.R.; Kim, S.Y.; Yoon, S.R.; Bae, K.; Jung, K.H. Substitution of Pro206 and Ser86 residues in the retinal binding pocket of Anabaena sensory rhodopsin is not sufficient for proton pumping function. J. Microbiol. Biotechnol. 2007, 17, 138–145. [Google Scholar]
- Jung, K.H.; Spudich, J.L. Protonatable residues at the cytoplasmic end of transmembrane helix-2 in the signal transducer HtrI control photochemistry and function of sensory rhodopsin I. Proc. Natl Acad. Sci. USA 1996, 93, 6557–6561. [Google Scholar]
- Jung, K.H.; Spudich, E.N.; Trivedi, V.D.; Spudich, J.L. An archaeal photosignal-transducing module mediates phototaxis in Escherichia coli. J. Bacteriol. 2001, 183, 6365–6371. [Google Scholar]
- Shimono, K.; Furutani, Y.; Kamo, N.; Kandori, H. Vibrational modes of the schiff base in pharaonis phoborhodopsin. Biochemistry 2003, 42, 7801–7806. [Google Scholar]
- Shimono, K.; Ikeura, Y.; Sudo, Y.; Iwamoto, M.; Kamo, N. Environment around the chromophore in pharaonis phoborhodopsin: mutation analysis of the retinal binding site. Biochim. Biophys. Acta 2001, 1515, 92–100. [Google Scholar]









































© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kawanabe, A.; Kandori, H. Photoreactions and Structural Changes of Anabaena Sensory Rhodopsin. Sensors 2009, 9, 9741-9804. https://doi.org/10.3390/s91209741
Kawanabe A, Kandori H. Photoreactions and Structural Changes of Anabaena Sensory Rhodopsin. Sensors. 2009; 9(12):9741-9804. https://doi.org/10.3390/s91209741
Chicago/Turabian StyleKawanabe, Akira, and Hideki Kandori. 2009. "Photoreactions and Structural Changes of Anabaena Sensory Rhodopsin" Sensors 9, no. 12: 9741-9804. https://doi.org/10.3390/s91209741
APA StyleKawanabe, A., & Kandori, H. (2009). Photoreactions and Structural Changes of Anabaena Sensory Rhodopsin. Sensors, 9(12), 9741-9804. https://doi.org/10.3390/s91209741