
The Phosphorylation of PDX-1 by Protein Kinase CK2 Is Crucial for Its Stability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
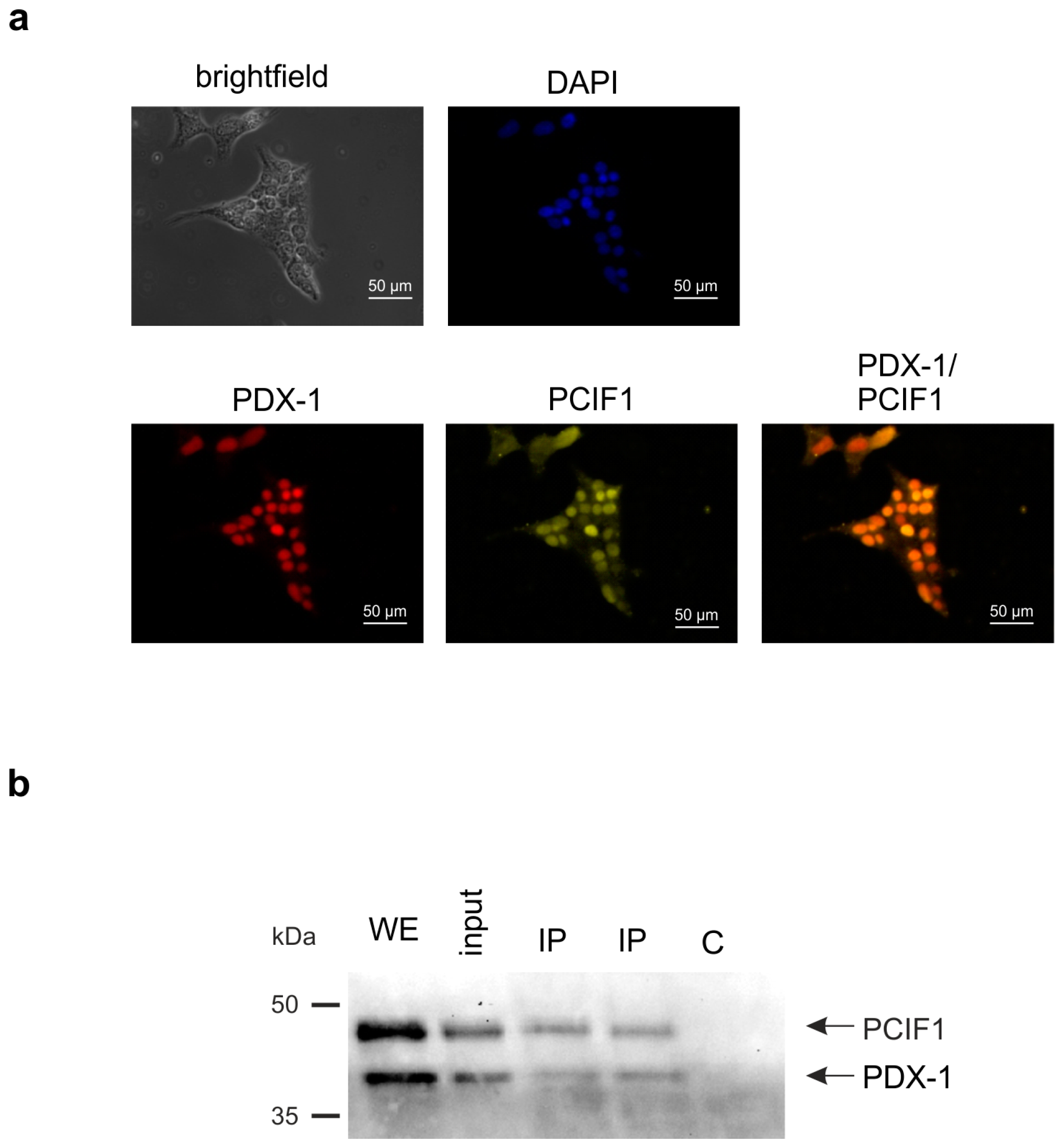
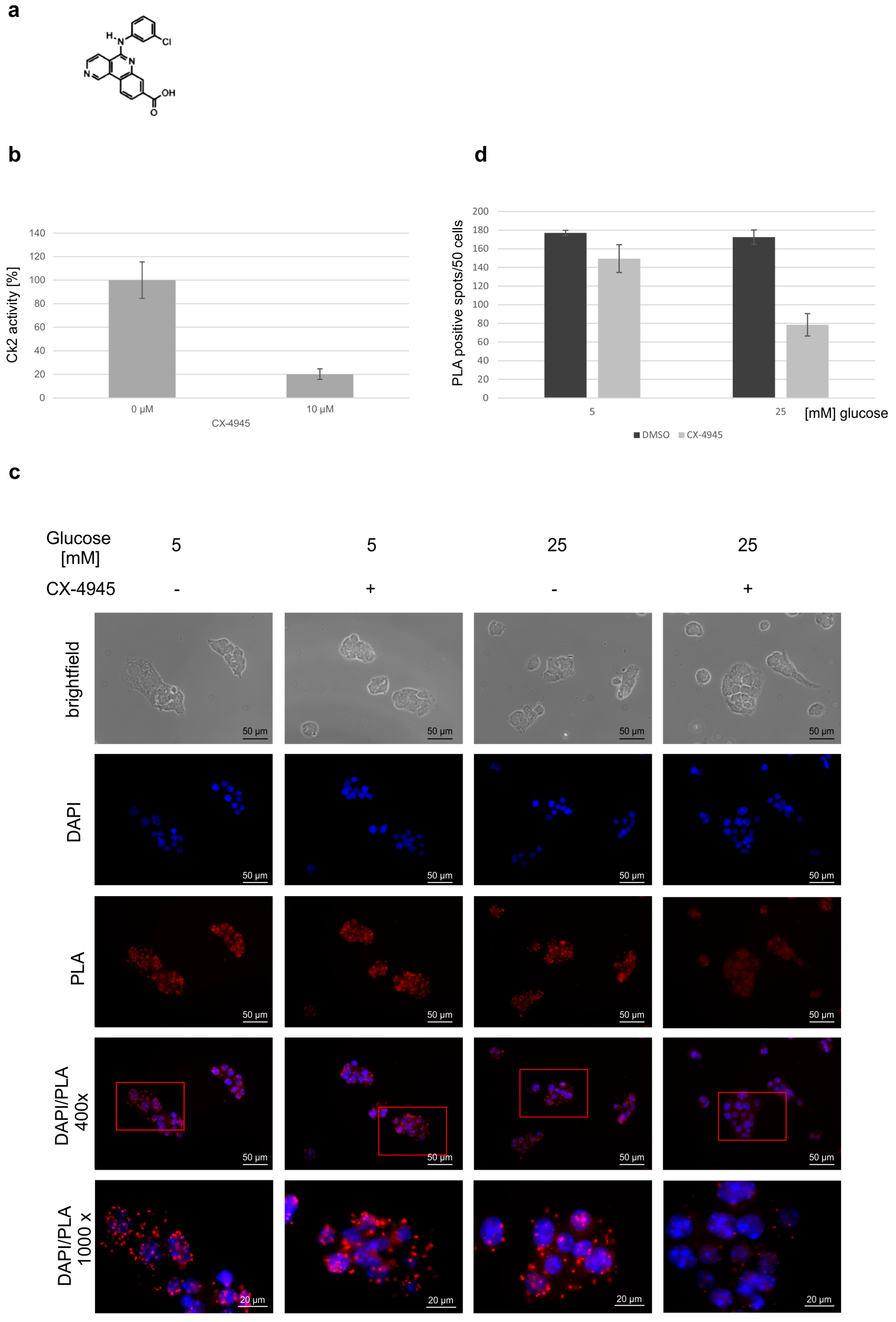
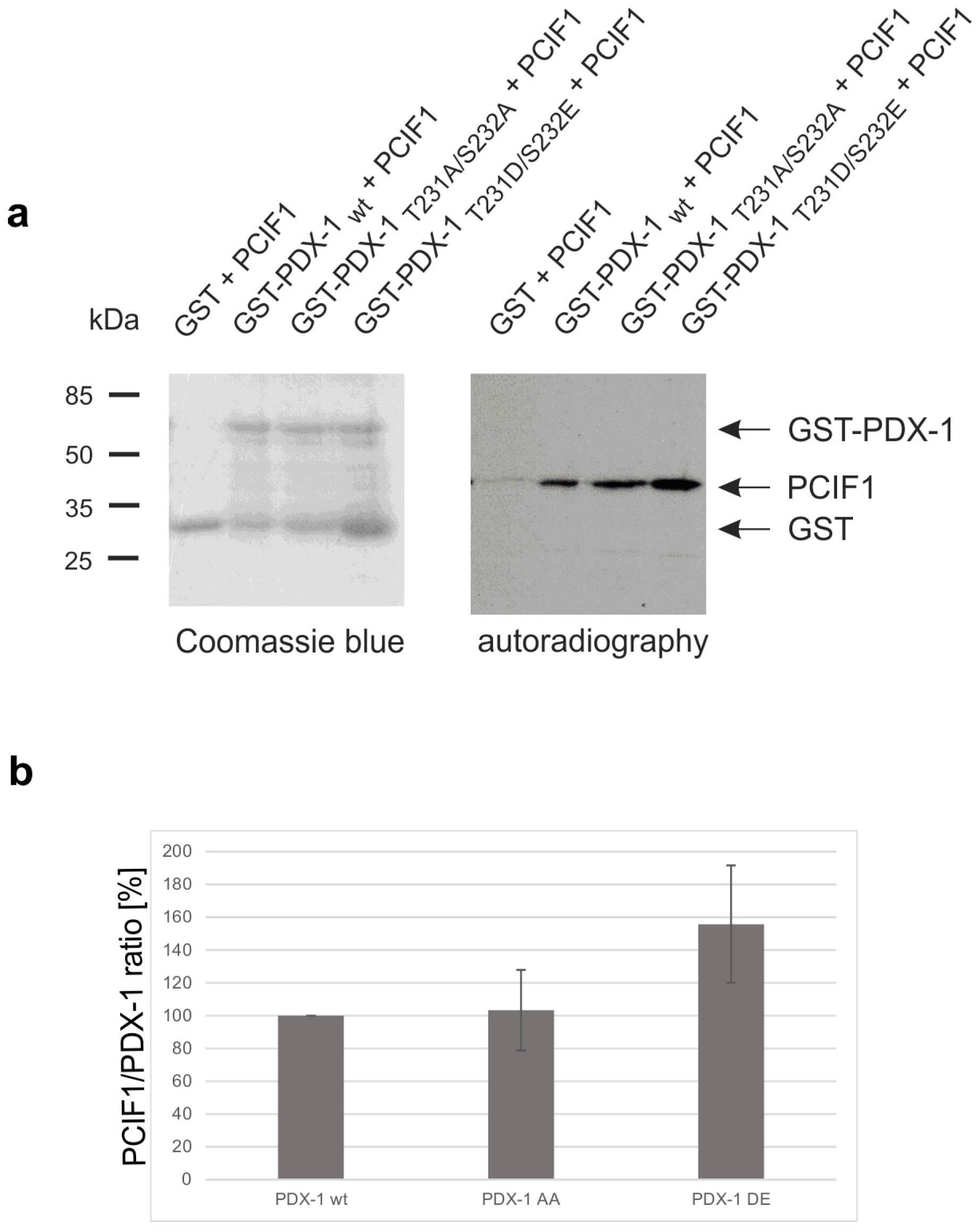
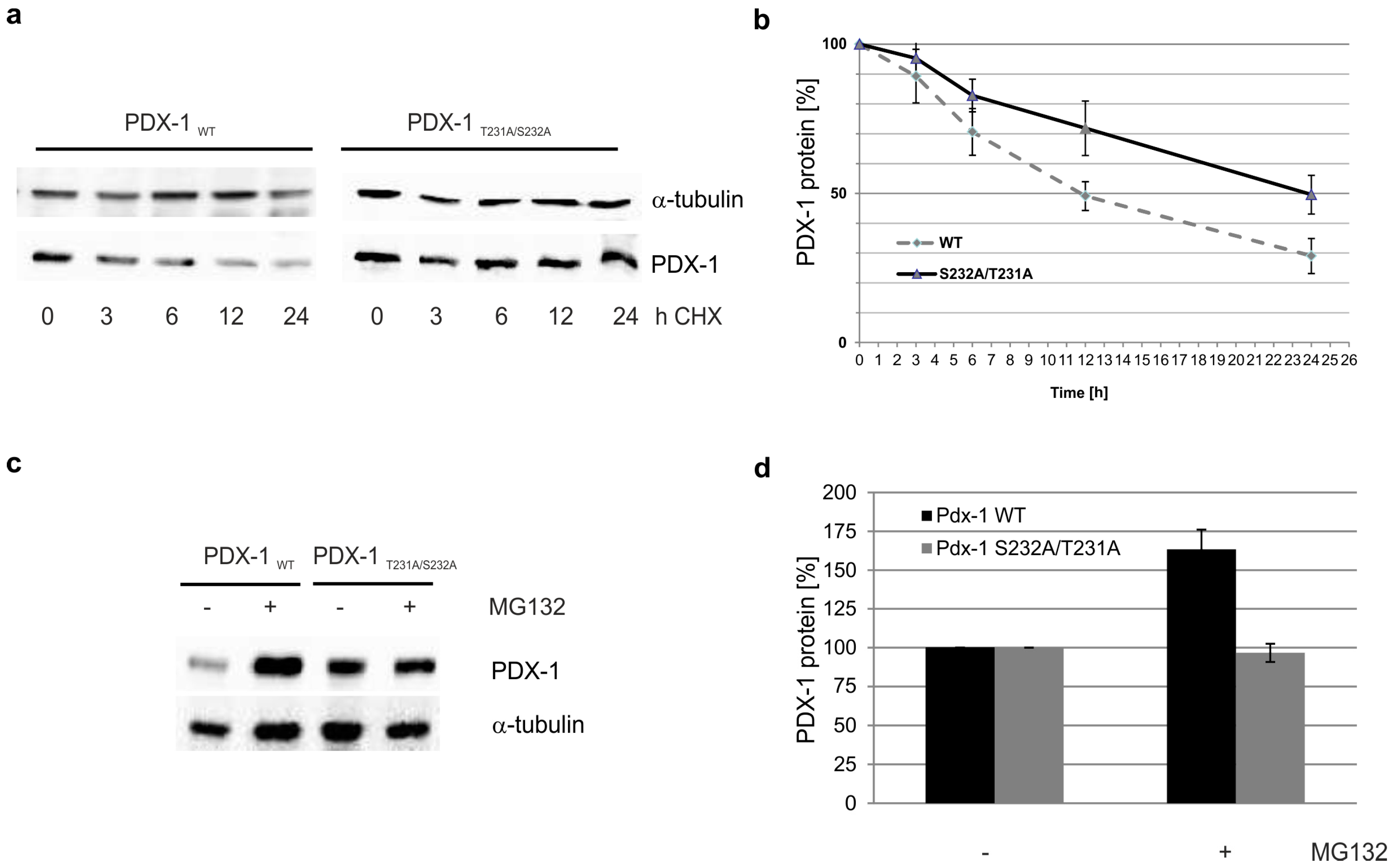
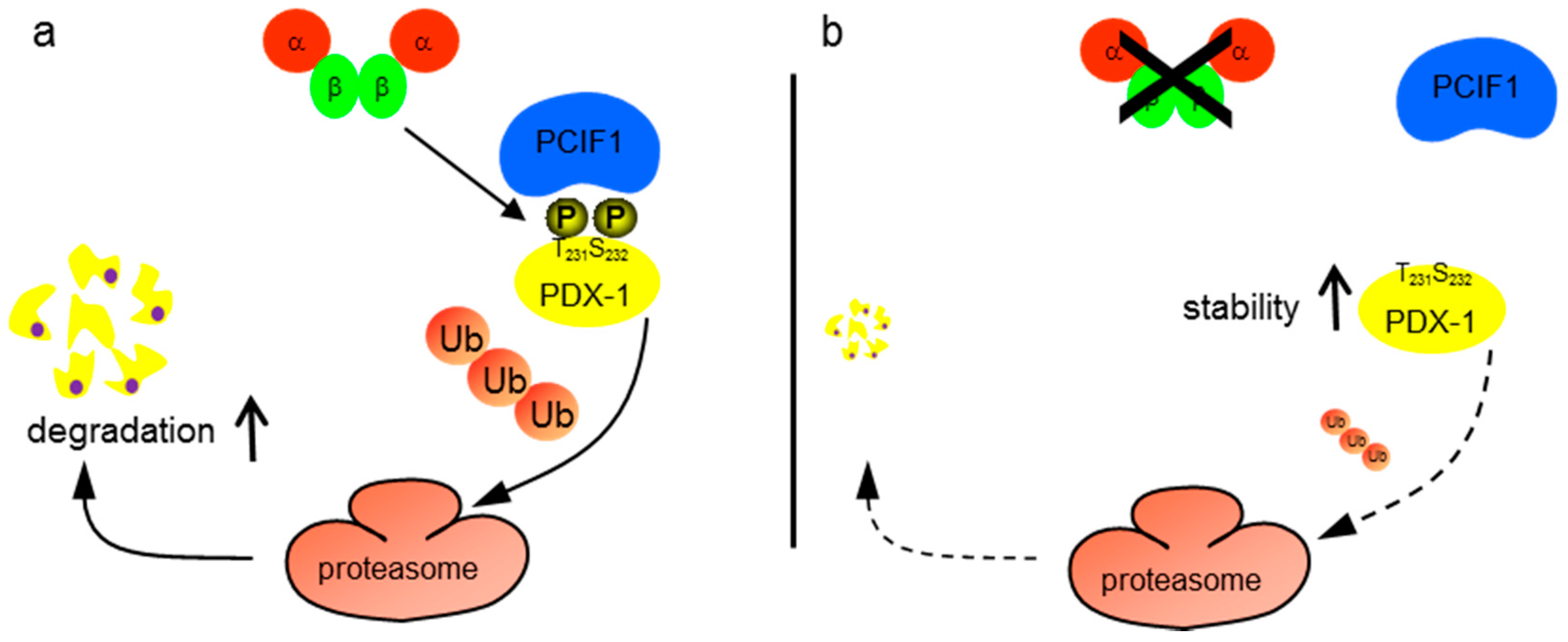
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Plasmids
4.3. Extraction of Cells and Western Blot Analysis
4.4. Co-Immunoprecipitation
4.5. Cycloheximide Chase
4.6. Immunofluorescence Analysis
4.7. Duolink® in Situ Proximity Ligation Assay
4.8. GST-Pull Down Assay
4.9. In Vitro Phosphorylation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.A.; Litchfield, D.W. From birth to death: The role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol. Life Sci. 2009, 66, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. CK2: A key player in cancer biology. Cell Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Osen-Sand, A.; Montessuit, S.; Magnenat, E.; Vilbois, F.; Hochmann, A.; Journot, L.; Antonsson, B.; Martinou, J.C. Phosphorylation of Bid by casein kinases I and II regulates its cleavage by caspase 8. Mol. Cell 2001, 8, 601–611. [Google Scholar] [CrossRef]
- Krippner-Heidenreich, A.; Talanian, R.V.; Sekul, R.; Kraft, R.; Thole, H.; Ottleben, H.; Luscher, B. Targeting of the transcription factor Max during apoptosis: phosphorylation-regulated cleavage by caspase-5 at an unusual glutamic acid residue in position P1. Biochem. J. 2001, 358, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Channavajhala, P.; Seldin, D.C. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene 2002, 21, 5280–5288. [Google Scholar] [CrossRef] [PubMed]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Grossman, S.R.; Takahashi, Y.; Rokas, M.V.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J. Biol. Chem. 2001, 276, 48627–48630. [Google Scholar] [PubMed]
- Ampofo, E.; Kietzmann, T.; Zimmer, A.; Jakupovic, M.; Montenarh, M.; Götz, C. Phosphorylation of the von Hippel-Lindau protein (VHL) by protein kinase CK2 reduces its protein stability and affects p53 and HIF-1α mediated transcription. Int. J. Biochem. Cell Biol. 2010, 42, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Al-Quobaili, F.; Müller, I.; Götz, C.; Thiel, G.; Montenarh, M. CK2 phosphorylation of PDX-1 regulates its transcription factor activity. Cell Mol. Life Sci. 2010, 67, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, S.; Brunicardi, F.C.; Wang, X.P. PDX-1 and the pancreas. Pancreas 2004, 28, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D. Transcription factors in islet development and physiology: Role of PDX-1 in beta-cell function. Ann. N. Y. Acad. Sci. 2004, 1014, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Moede, T.; Leibiger, B.; Pour, H.G.; Berggren, P.; Leibiger, I.B. Identification of a nuclear localization signal, RRMKWKK, in the homeodomain transcription factor PDX-1. FEBS Lett. 1999, 461, 229–234. [Google Scholar] [CrossRef]
- Hessabi, B.; Ziegler, P.; Schmidt, I.; Hessabi, C.; Walther, R. The nuclear localization signal (NLS) of PDX-1 is part of the homeodomain and represents a novel type of NLS. Eur. J. Biochem. 1999, 263, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Hani, E.H.; Stoffers, D.A.; Chevre, J.C.; Durand, E.; Stanojevic, V.; Dina, C.; Habener, J.F.; Froguel, P. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J. Clin. Invest. 1999, 104, R41–R48. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, B.N.; Bermano, G.; Boodram, L.L.; Teelucksingh, S.; Tsuchiya, T.; Mahabir, D.; Allan, A.B.; Stein, R.; Docherty, K.; et al. Insulin promoter factor-1 mutations and diabetes in Trinidad: Identification of a novel diabetes-associated mutation (E224K) in an Indo-Trinidadian family. J. Clin. Endocrinol. Metab. 2004, 89, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Miller, C.; Habener, J.F. Functional regions of the homeodomain protein IDX-1 required for transactivation of the rat somatostatin gene. Endocrinology 1996, 137, 2959–2967. [Google Scholar] [PubMed]
- Al-Quobaili, F.; Montenarh, M. Pancreatic duodenal homeobox factor-1 and diabetes mellitus type 2. Int. J. Mol. Med. 2008, 21, 399–404. [Google Scholar] [PubMed]
- Liu, A.; Desai, B.M.; Stoffers, D.A. Identification of PCIF1, a POZ domain protein that inhibits PDX-1 (MODY4) transcriptional activity. Mol. Cell Biol. 2004, 24, 4372–4383. [Google Scholar] [CrossRef] [PubMed]
- Claiborn, K.C.; Sachdeva, M.M.; Cannon, C.E.; Groff, D.N.; Singer, J.D.; Stoffers, D.A. PCIF1 modulates PDX1 protein stability and pancreatic beta cell function and survival in mice. J. Clin. Invest. 2010, 120, 3713–3721. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today 2014, 19, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, O.; Leuchowies, K.J.; Gullberg, M.; Janoius, M.; Weibrecht, O.; Larsson, L.G.; Landegren, U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 2008, 45, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O'Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Fang, Z.; Yu, X.; Zhang, M. Transcription factors involved in glucose-stimulated insulin secretion of pancreatic beta cells. Biochem. Biophys. Res. Commun. 2009, 384, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Einarson, M.B.; Pugacheva, E.N.; Orlinick, J.R. GST Pull-down. CSH Protoc. 2007. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Marin, O.; Pinna, L.A. Substrate specificity of protein kinase CK2. Cell Mol. Biol. Res. 1994, 40, 401–409. [Google Scholar] [PubMed]
- Humphrey, R.K.; Yu, S.M.; Flores, L.E.; Jhala, U.S. Glucose regulates steady-state levels of PDX1 via the reciprocal actions of GSK3 and AKT kinases. J. Biol. Chem. 2010, 285, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Selander, L.; Carlsson, L.; Edlund, H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J. Biol. Chem. 2006, 281, 6395–6403. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Breuer, P.; Schmitt, I.; Walter, J.; Evert, B.O.; Wüllner, U. CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3. Hum. Mol. Genet. 2009, 18, 3334–3343. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Tan, M.; Pamarthy, D.; Wang, G.; Ahmed, K.; Sun, Y. CK2 phosphorylation of SAG at Thr10 regulates SAG stability, but not its E3 ligase activity. Mol. Cell Biochem. 2007, 295, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guan, B.; Maghami, S.; Bieberich, C.J. NKX3.1 is regulated by protein kinase CK2 in prostate tumor cells. Mol. Cell Biol. 2006, 26, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, P.; Montminy, M.R.; Van, O.E. Regulation of the pancreatic duodenal homeobox-1 protein by DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 38203–38210. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- An, R.; da Silva Xavier, G.; Semplici, F.; Vakhshouri, S.; Hao, H.X.; Rutter, J.; Pagano, M.A.; Meggio, F.; Pinna, L.A.; Rutter, G.A. Pancreatic and duodenal homeobox 1 (PDX1) phosphorylation at serine-269 is HIPK2-dependent and affects PDX1 subnuclear localization. Biochem. Biophys. Res. Commun. 2010, 399, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, H.; Liu, S.H.; Shahi, K.M.; Lin, X.; Wu, J.; Feng, X.H.; Qin, J.; Tan, T.H.; Brunicardi, F.C. p38 MAP kinase interacts with and stabilizes pancreatic and duodenal homeobox-1. Curr. Mol. Med. 2013, 13, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-substrate complexes: Insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ao, J.; Fu, J.; Lee, D.F.; Xu, J.; Lonard, D.; O'Malley, B.W. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene 2011, 30, 4350–4364. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Shukla, N.; Thacker, G.; Kapoor, I.; Lochab, S.; Bhatt, M.L.; Chattopadhyay, N.; Sanyal, S.; Trivedi, A.K. Ubiquitin ligase, Fbw7, targets CDX2 for degradation via two phosphodegron motifs in a GSK3β-dependent manner. Mol. Cancer Res. 2016, 14, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, S.; Carreira, S.; Bailey, D.; Abaitua, F.; O'Hare, P. Phosphorylation and SCF-mediated degradation regulate CREB-H transcription of metabolic targets. Mol. Biol. Cell 2015, 26, 2939–2954. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Gao, W.W.; Tang, H.M.; Deng, J.J.; Wong, C.M.; Chan, C.P.; Jin, D.Y. β-TrCP-mediated ubiquitination and degradation of liver-enriched transcription factor CREB-H. Sci. Rep. 2016, 6, 23938. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Dai, X.; Lunardi, A.; Li, Z.; Inuzuka, H.; Liu, P.; Varmeh, S.; Zhang, J.; Cheng, L.; Sun, Y.; et al. SPOP promotes ubiquitination and degradation of the ERG oncoprotein to suppress prostate cancer progression. Mol. Cell 2015, 59, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Bunce, M.W.; Boronenkov, I.V.; Anderson, R.A. Coordinated activation of the nuclear ubiquitin ligase Cul3-SPOP by the generation of phosphatidylinositol 5-phoshpate. J. Biol. Chem. 2008, 283, 8678–8686. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Miyazaki, J.; Araki, K.; Yamato, E.; Ikegami, H.; Asano, T.; Shibasaki, Y.; Oka, Y.; Yamamura, K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology 1990, 127, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; La, M.; Oh, K.H.; Oh, Y.M.; Kim, G.R.; Seol, J.H.; Baek, S.H.; Chiba, T.; Tanaka, K.; Bang, O.S.; et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J. Biol. Chem. 2006, 281, 12664–12672. [Google Scholar] [CrossRef] [PubMed]
- Faust, M.; Schuster, N.; Montenarh, M. Specific binding of protein kinase CK2 catalytic subunits to tubulin. FEBS Lett. 1999, 462, 51–56. [Google Scholar] [CrossRef]
- Sun, Q.; Yu, X.; Degraff, D.J.; Matusik, R.J. Upstream stimulatory factor 2, a novel FoxA1-interacting protein, is involved in prostate-specific gene expression. Mol. Endocrinol. 2009, 23, 2038–2047. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, S.; Meng, R.; Montenarh, M.; Götz, C. The Phosphorylation of PDX-1 by Protein Kinase CK2 Is Crucial for Its Stability. Pharmaceuticals 2017, 10, 2. https://doi.org/10.3390/ph10010002
Klein S, Meng R, Montenarh M, Götz C. The Phosphorylation of PDX-1 by Protein Kinase CK2 Is Crucial for Its Stability. Pharmaceuticals. 2017; 10(1):2. https://doi.org/10.3390/ph10010002
Chicago/Turabian StyleKlein, Sabrina, Rui Meng, Mathias Montenarh, and Claudia Götz. 2017. "The Phosphorylation of PDX-1 by Protein Kinase CK2 Is Crucial for Its Stability" Pharmaceuticals 10, no. 1: 2. https://doi.org/10.3390/ph10010002
APA StyleKlein, S., Meng, R., Montenarh, M., & Götz, C. (2017). The Phosphorylation of PDX-1 by Protein Kinase CK2 Is Crucial for Its Stability. Pharmaceuticals, 10(1), 2. https://doi.org/10.3390/ph10010002