
Systematic Analysis of Pharmaceutical Preparations of Chondroitin Sulfate Combined with Glucosamine

 and
and
Abstract
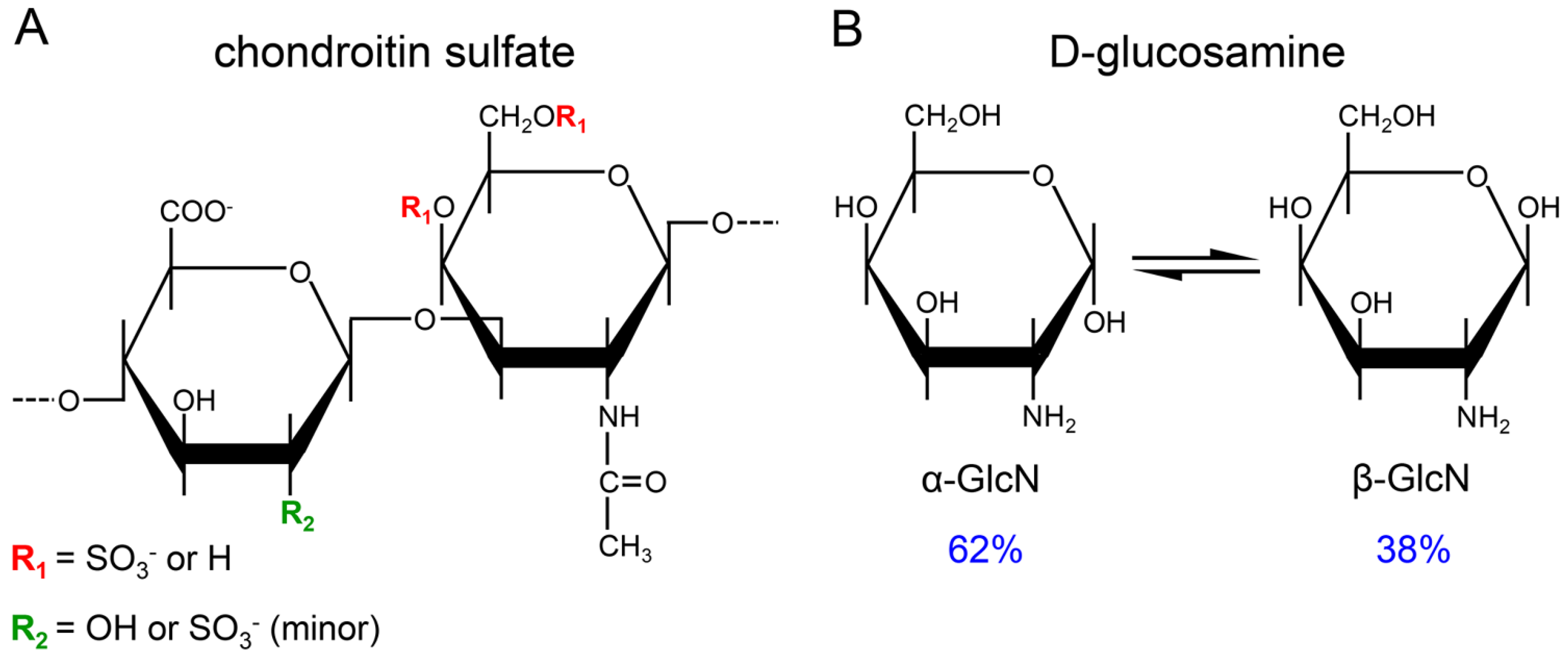
:1. Introduction
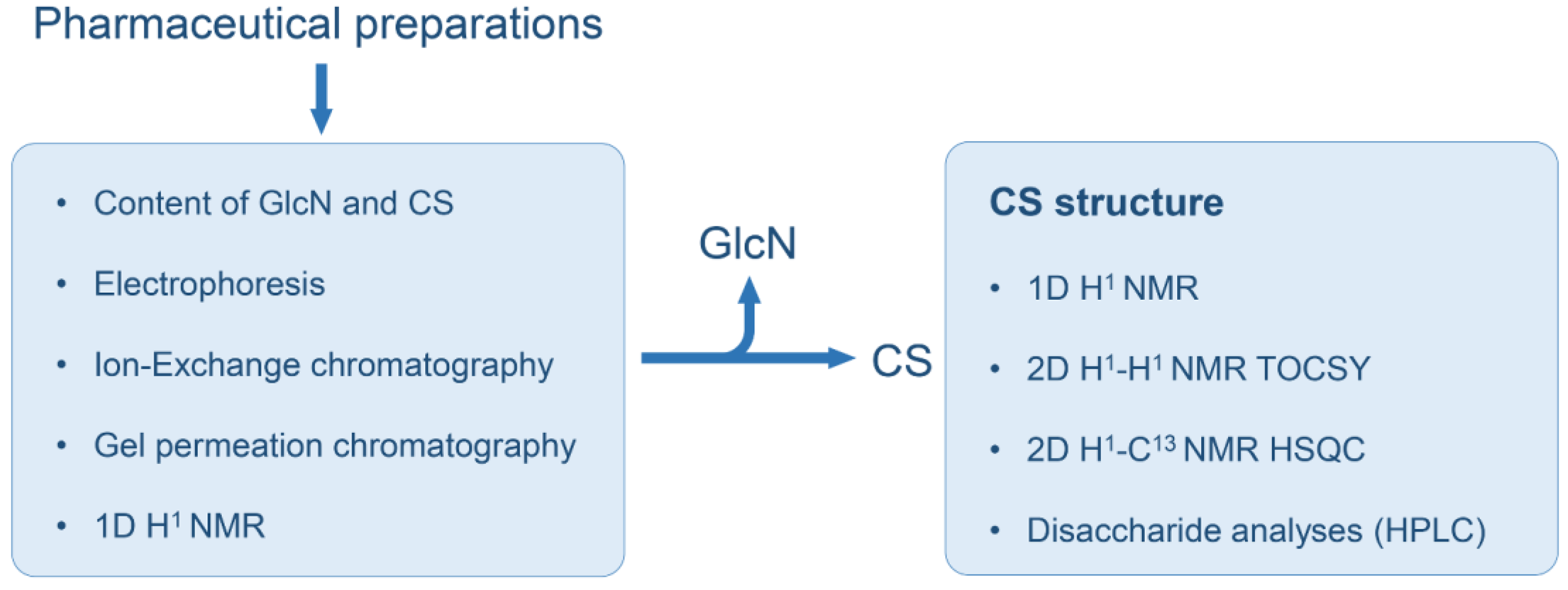
2. Materials and Methods
2.1. Pharmaceutical Formulations of CS Combined with GlcN and CS Standards
2.2. Chemical Analyses of the CS and GlcN Contents
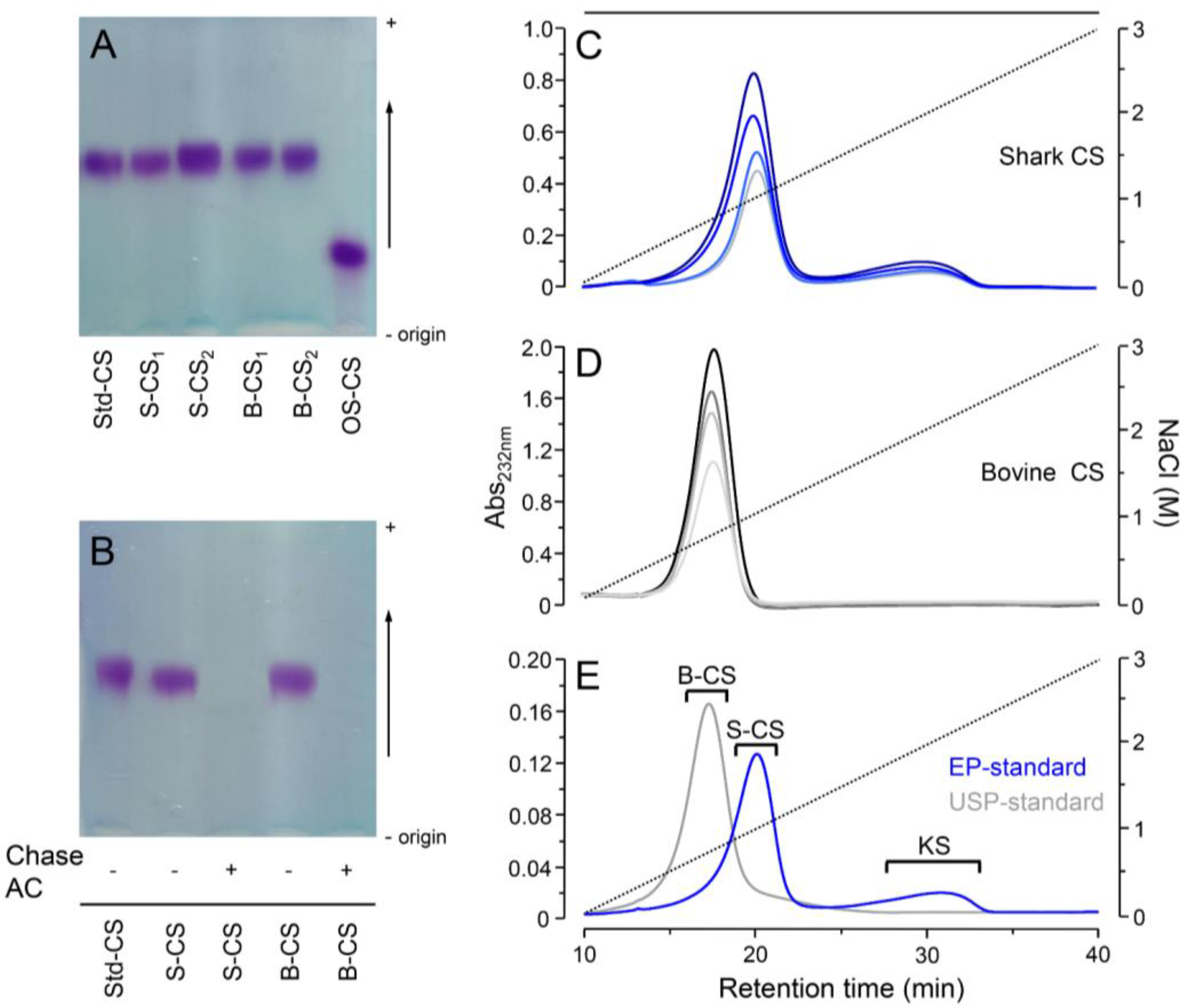
2.3. Agarose-Gel Electrophoresis
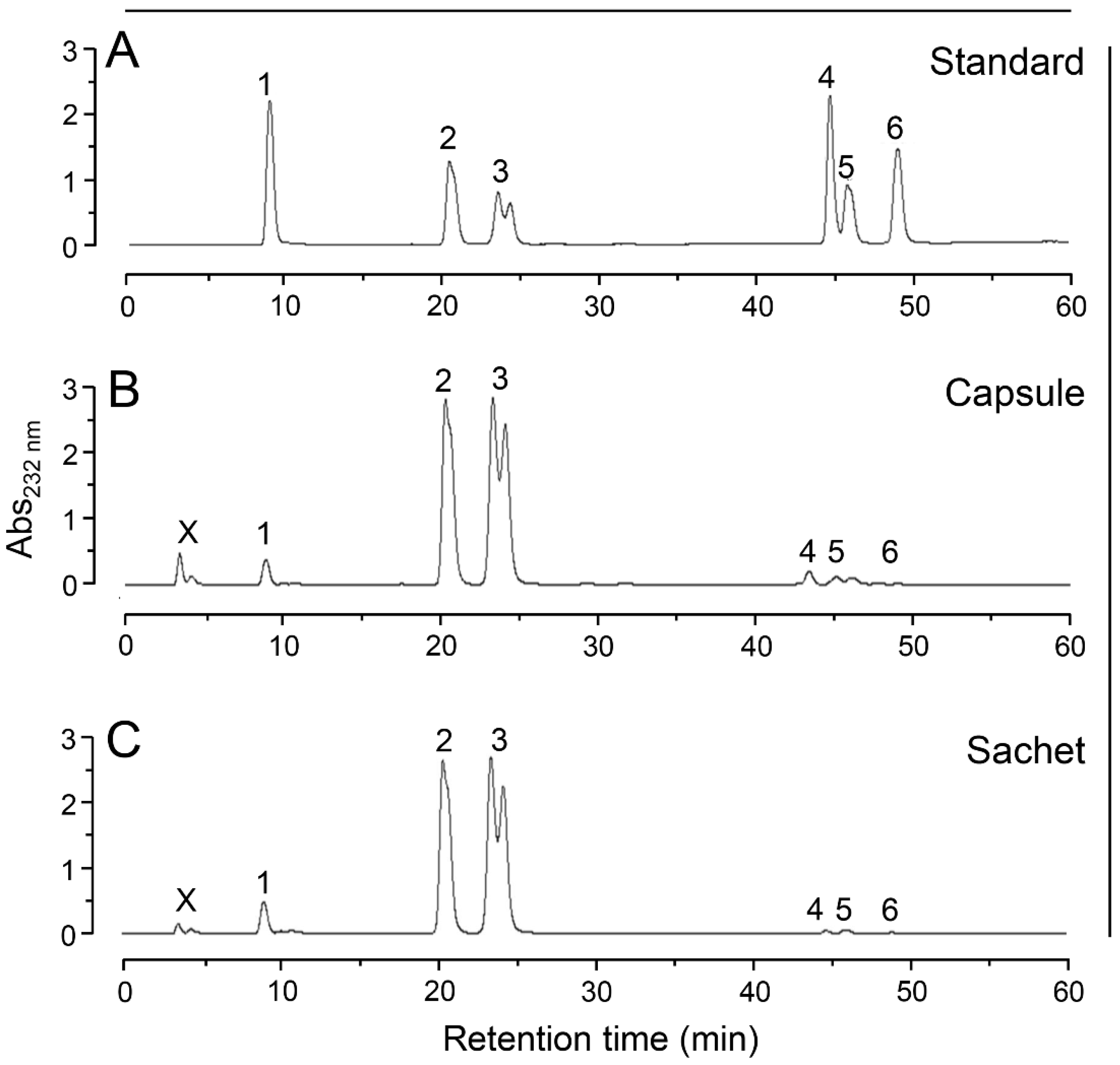
2.4. Anion-Exchange Chromatography
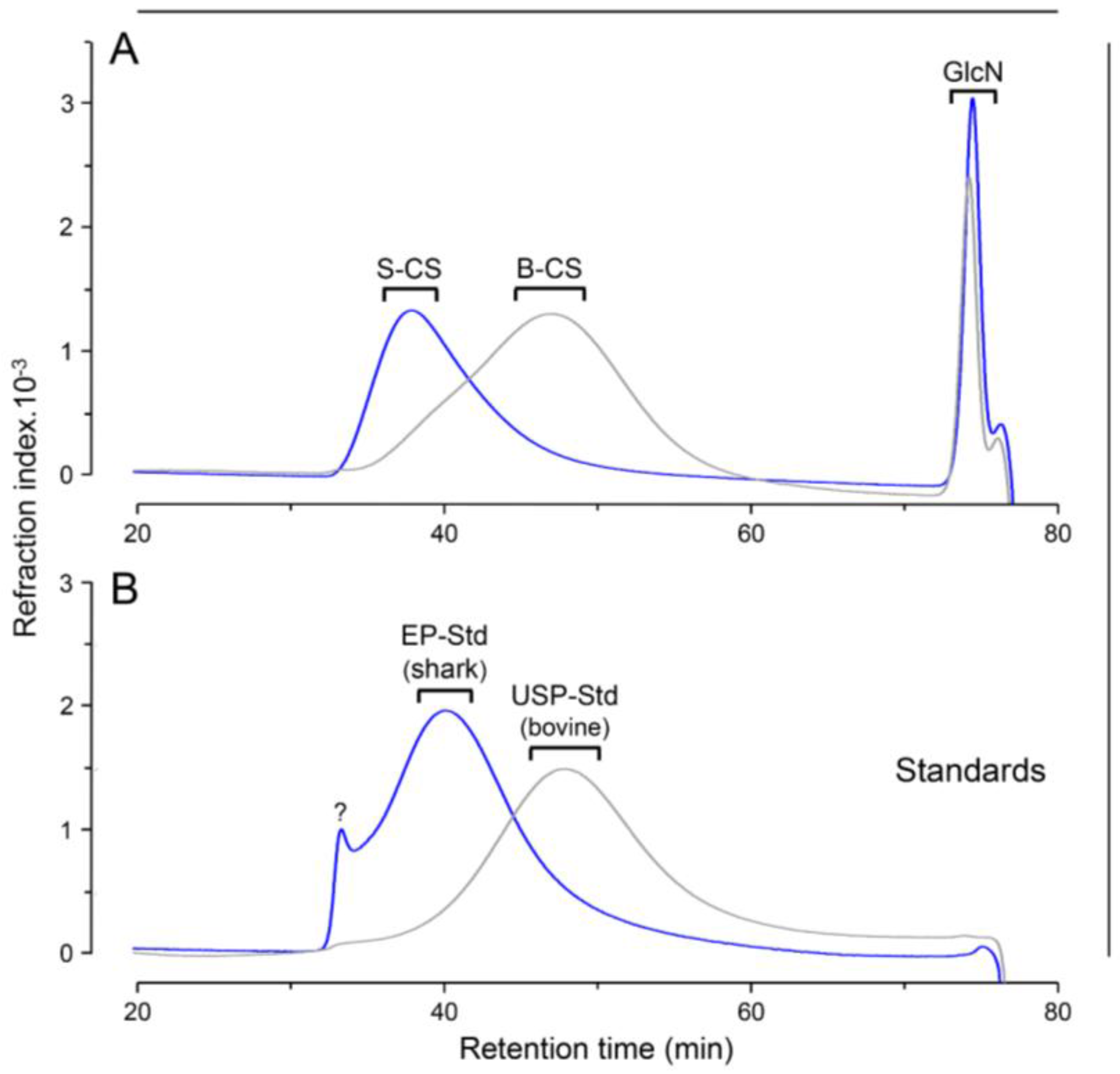
2.5. Size-Exclusion Chromatography
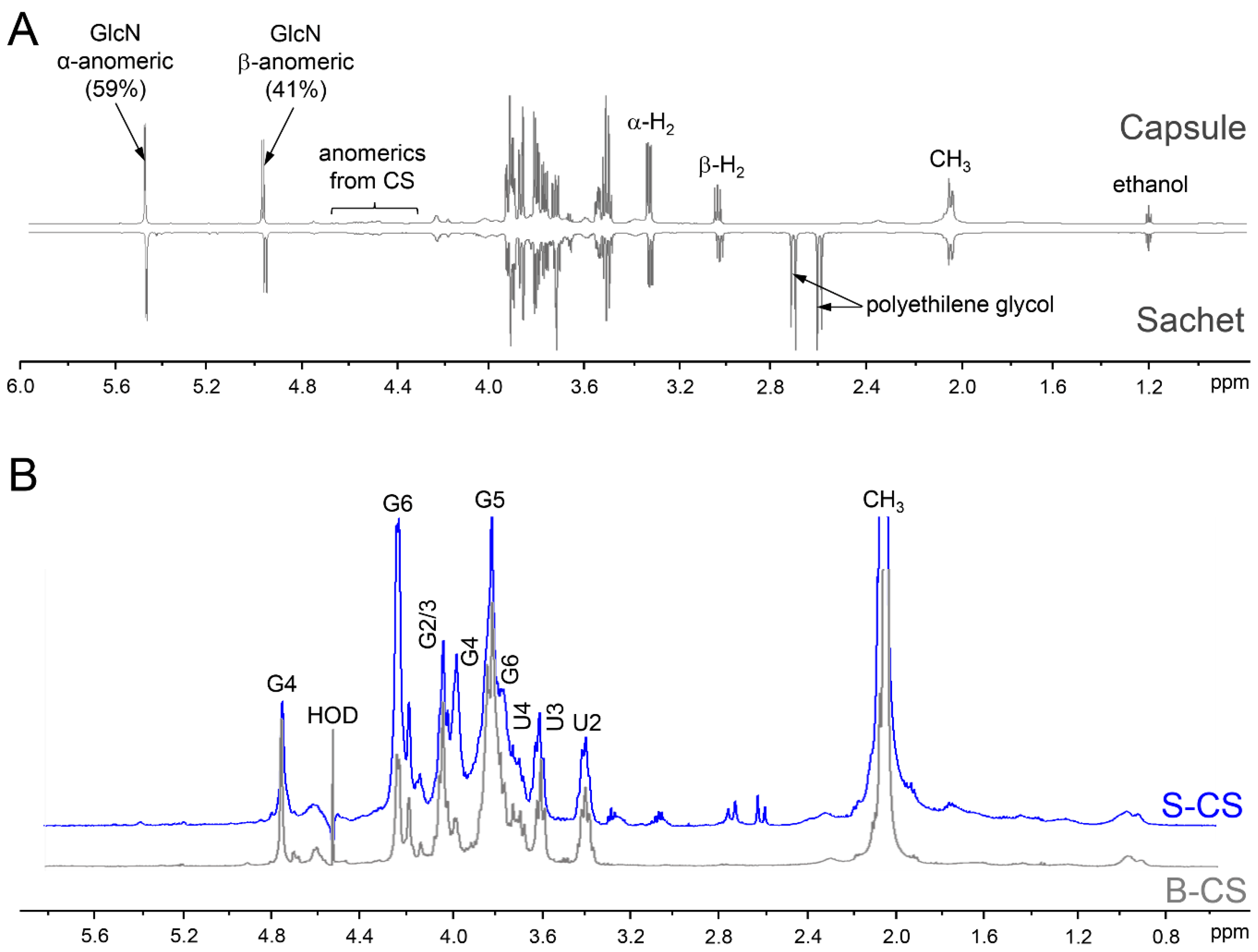
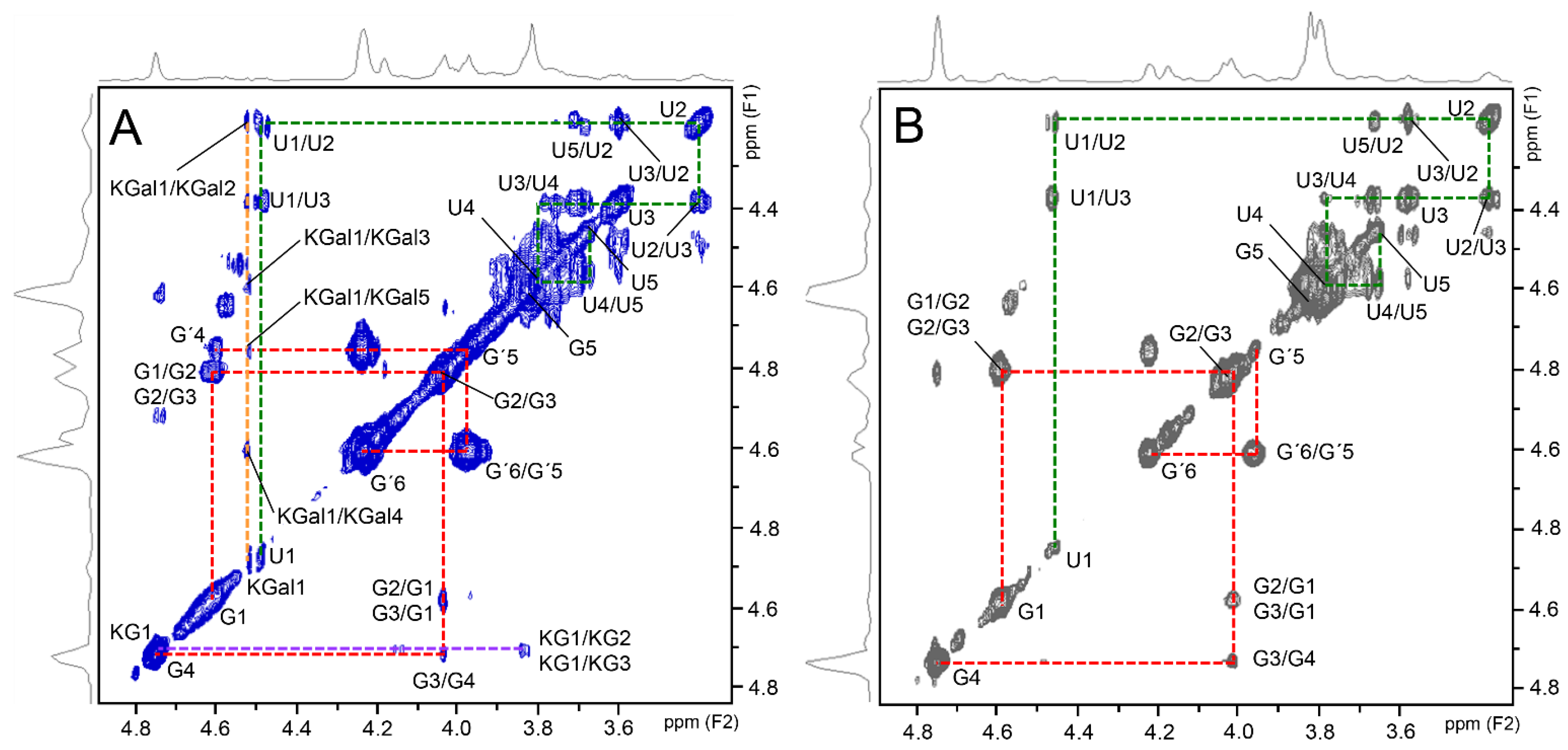
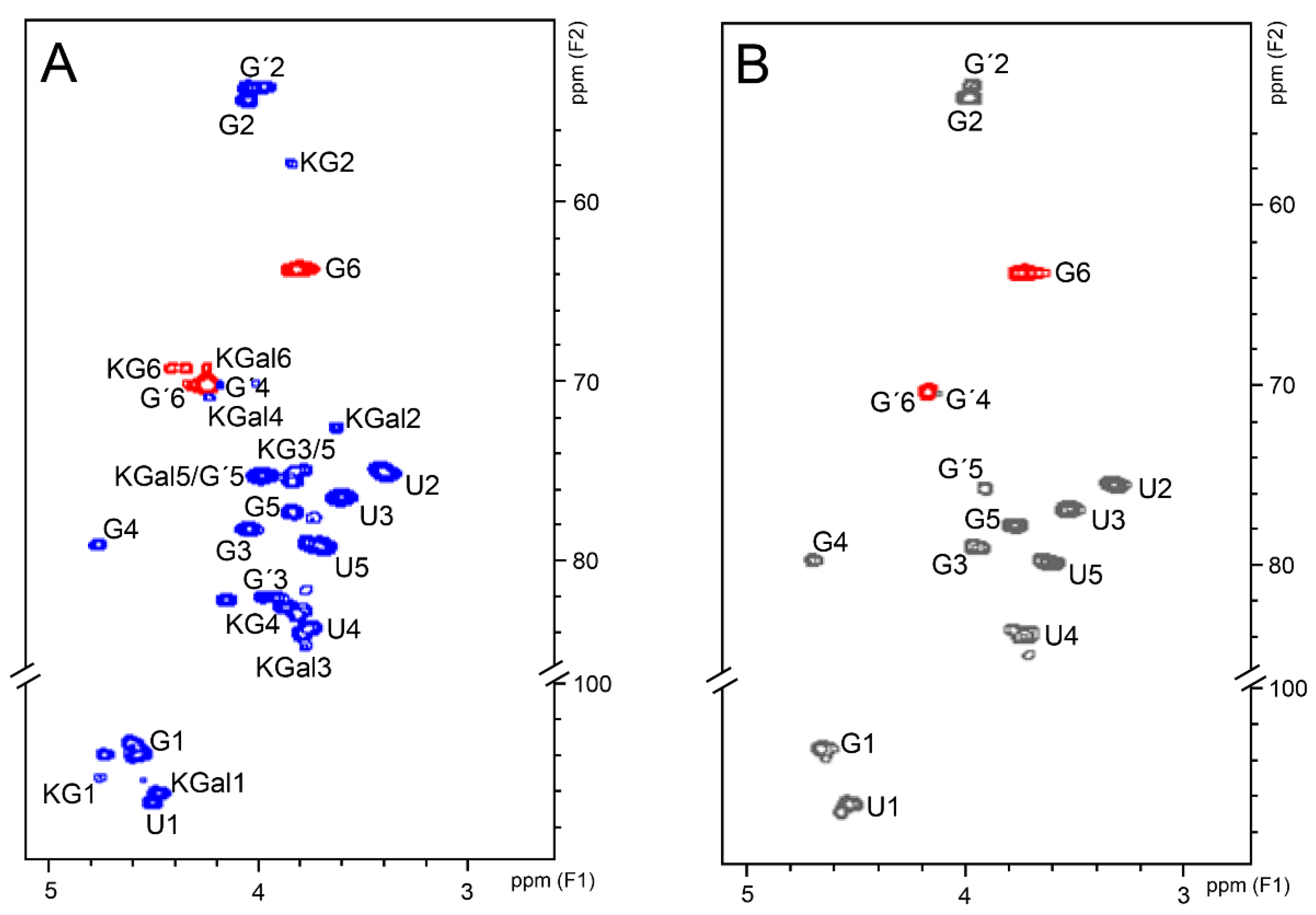
2.6. NMR Analyses
2.7. Analysis of the Disaccharide Composition
3. Results and Discussion
4. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of heparin and related drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Tovar, A.M.; Santos, G.R.; Capillé, N.V.; Piquet, A.A.; Glauser, B.F.; Pereira, M.S.; Vilanova, E.; Mourão, P.A. Structural and haemostatic features of pharmaceutical heparins from different animal sources: Challenges to define thresholds separating distinct drugs. Sci. Rep. 2016, 6, 35619. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, M.; Jain, A.; Hurkat, P.; Jain, S.K. Chondroitin sulphate: A focus on osteoarthritis. Glycoconj. J. 2016, 5, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Garnjanagoonchorn, W.; Wongekalak, L.; Engkagul, A. Determination of chondroitin sulfate from different sources of cartilage. Chem. Eng. Process. 2007, 46, 465–471. [Google Scholar] [CrossRef]
- Pomin, V.H.; Piquet, A.A.; Pereira, M.S.; Mourão, P.A. Residual keratan sulfate in chondroitin sulfate formulations for oral administration. Carbohydr. Polym. 2012, 90, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ozimek, L. Detection of keratan sulfate by immunological methods in commercial chondroitin sulfate preparations. Carbohydr. Polym. 2014, 99, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Restaino, O.F.; Finamore, R.; Diana, P.; Marseglia, M.; Vitiello, M.; Casillo, A.; Bedini, E.; Parrilli, M.; Corsaro, M.M.; Trifuoggi, M.; et al. A multi-analytical approach to better assess the keratan sulfate contamination in animal origin chondroitin sulfate. Anal. Chim. Acta 2017, 958, 59–70. [Google Scholar] [CrossRef] [PubMed]
- European Pharmacopeia. Chondroitin sulfate sodium monograph. In European Pharmacopeia, 7th ed.; European Directorate for the Quality of Medicines: Strasbourg, France, 2007; pp. 1681–1683. [Google Scholar]
- United States Pharmacopoeia. Chondroitin sulfate sodium monograph [Revision Bulletin, 2015]. In United States Pharmacopoeia, 39th ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2016; pp. 1–3. [Google Scholar]
- Reginster, J.Y.; Neuprez, A.; Lecart, M.P.; Sarlet, N.; Bruyere, O. Role of glucosamine in the treatment for osteoarthritis. Rheumatol. Int. 2012, 10, 2959–2967. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Mobasheri, A.; Marty, M. Is there any scientific evidence for the use of glucosamine in the management of human osteoarthritis? Arthritis Res. Ther. 2012, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.O.; Reda, D.J.; Harris, C.L.; Klein, M.A.; O’Dell, J.R.; Hooper, M.M.; Bradley, J.D.; Bingham, C.O., 3rd; Weisman, M.H.; Jackson, C.G.; et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N. Engl. J. Med. 2006, 354, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Mathy, M.; Sanchez, C.; Lambert, C. Chondroitin sulfate in the treatment of osteoarthritis: From in vitro studies to clinical recommendations. Ther. Adv. Musculoskelet. Dis. 2010, 6, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.M.; Avenoso, A.; Campo, S.; D’ascola, A.; Traina, P.; Sama, D.; Calatroni, A. Glycosaminoglycans modulate inflammation and apoptosis in LPS-treated chondrocytes. J. Cell. Biochem. 2009, 106, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.M.; Avenoso, A.; Campo, S.; Traina, P.; D’ascola, A.; Calatroni, A. Glycosaminoglycans reduced inflammatory response by modulating toll-like receptor-4 in LPS-stimulated chondrocytes. Arch. Biochem. Biophys. 2009, 497, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Kaplun, M.; Williams, D.Y.; Xu, D.; Ateshian, G.A.; Hung, C.T. Influence of chondroitin sulfate on the biochemical, mechanical and frictional properties of cartilage explants in longterm culture. J. Biomech. 2009, 42, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Imada, K.; Oka, H.; Kawasaki, D.; Miura, N.; Sato, T.; Ito, A. Anti-arthritic action mechanisms of natural chondroitin sulfate in human articular chondrocytes and synovial fibroblasts. Biol. Pharm. Bull. 2010, 33, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Sim, J.S.; Jeong, C.S.; Chang, S.Y.; Choi, D.W.; Toida, T.; Kim, Y.S. Effects of low molecular weight chondroitin sulfate on type II collagen-induced arthritis in DBA/1J mice. Biol. Pharm. Bull. 2004, 27, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.M.; Vergnolle, N.; McDougall, J.J.; Wallace, J.L.; Marty, S.; Teskey, V.; Buret, A.G. Effects of chondroitin and glucosamine sulfate in a dietary bar formulation on inflammation, interleukin-1β, matrix metalloprotease-9, and cartilage damage in arthritis. Exp. Biol. Med. 2005, 230, 255–262. [Google Scholar]
- Campo, G.M.; Avenoso, A.; Campo, S.; D’ascola, A.; Traina, P.; Calatroni, A. Chondroitin-4-sulphate inhibits NF-kB translocation and caspase activation in collagen-induced arthritis in mice. Osteoarthr. Cartil. 2008, 16, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Wandel, S.; Jüni, P.; Tendal, B.; Nüesch, E.; Villiger, P.M.; Welton, N.J.; Reichenbach, S.; Trelle, S. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: Network meta-analysis. BMJ 2010, 4, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Reginster, J.Y.; Deroisy, R.; Rovati, L.C.; Lee, R.L.; Lejeune, E.; Bruyere, O.; Giacovelli, G.; Henrotin, Y.; Dacre, J.E.; Gossett, C. Long-term effects of glucosamine sulphate on osteoarthritis progression: A randomised, placebo-controlled clinical trial. Lancet 2001, 357, 251–256. [Google Scholar] [CrossRef]
- McAlindon, T.; Formica, M.; LaValley, M.; Lehmer, M.; Kabbara, K. Effectiveness of glucosamine for symptoms of knee osteoarthritis: Results from an internet-based randomized double-bind controlled trial. Am. J. Med. 2004, 117, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.A.; Stucki, G.; Frey, D.; De Vathaire, F.; Vignon, E.; Bruehlmann, P.; Uebelhart, D. Chondroitins 4 and 6 sulfate in osteoarthritis of the knee: A randomized, controlled trial. Arthritis Rheum. 2005, 52, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Mazières, B.; Hucher, M.; Zaïm, M.; Garnero, P. Effect of chondroitin sulphate in symptomatic knee osteoarthritis: A multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2007, 66, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Gardell, S. Determination of Hexosamines. In Methods of Biochemical Analysis; Glick, D., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1958; Volume 6, pp. 256–288. [Google Scholar]
- Volpi, N. Analytical aspects of pharmaceutical grade chondroitin sulfates. J. Pharm. Sci. 2007, 12, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.R.; Kozlowski, E.O.; Cerqueira, V.R.; Castelo-Branco, M.T.; Costa, M.L.; Pavão, M.S. Chondroitin sulfate and keratan sulfate are the major glycosaminoglycans present in the adult zebrafish Danio rerio (Chordata-Cyprinidae). Glycoconj. J. 2007, 9, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Huckerby, T.N. The keratan sulphates: Structural investigations using NMR spectroscopy. Progr. Nucl. Magn. Reson. Spectrosc. 2002, 40, 35–110. [Google Scholar] [CrossRef]
- Vilanova, E.; Glauser, B.F.; Oliveira, S.M.; Tovar, A.M.; Mourão, P.A. Update on Brazilian biosimilar enoxaparins. Expert Rev. Hematol. 2016, 9, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Lauder, R.M.; Huckerby, T.N.; Nieduszynski, I.A. A fingerprinting method for chondroitin/dermatan sulfate and hyaluronan oligosaccharides. Glycobiology 2000, 10, 393–401. [Google Scholar] [CrossRef] [PubMed]
- De Waard, P.; Vliegenthart, J.F.; Harada, T.; Sugahara, K. Structural studies on sulfated oligosaccharides derived from the carbohydrate-protein linkage region of chondroitin 6-sulfate proteoglycans of shark cartilage. II. Seven compounds containing 2 or 3 sulfate residues. J. Biol. Chem. 1992, 267, 6036–6043. [Google Scholar] [PubMed]
- Chess, E.K.; Bairstow, S.; Donovan, S.; Havel, K.; Hu, P.; Johnson, R.J.; Lee, S.; McKee, J.; Miller, R.; Moore, E.; et al. Case study: Contamination of heparin with oversulfated chondroitin sulfate. Handb. Exp. Pharmacol. 2012, 207, 99–125. [Google Scholar]
- United States Pharmacopoeia. Glucosamine sulfate sodium chloride monograph. In United States Pharmacopoeia, 29th ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2006; p. 2342. [Google Scholar]
- Adebowale, A.; Cox, D.S.; Liang, Z.; Eddington, N.D. Analysis of Glucosamine and Chondroitin Sulfate Content in Marketed Products and the Caco-2 Permeability of Chondroitin Sulfate Raw Materials. J. Am. Nutraceut. Assoc. 2000, 3, 37–44. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Component | Declared Content (mg) | Observed Content (mg) a |
|---|---|---|---|
| Capsule | GlcN b | 500 | 465 ± 20 b |
| CS c | 400 | 376 ± 8 c | |
| Sachet | GlcN | 1500 | 1500 ± 60 b |
| CS | 1200 | 1211 ± 179 c |
| Signal | Chemical Shifts (ppm) | ||||
|---|---|---|---|---|---|
| CS | KS | ||||
| GalNAc-4S | GalNAc-6S | GlcA | GalNAc-6S | Gal-6S | |
| H1/C1 | 4.58/103.6 | 4.58/103.6 | 4.46/106.5 | 4.74/105.2 | 4.56/105.3 |
| H2/C2 | 4.02/54.0 | 4.02/53.4 | 3.37/75.2 | 3.85/57.6 | 3.62/72.3 |
| H3/C3 | 4.00/78.5 a | 3.98/81.7 | 3.57/76.2 | 3.83/75.3 | 3.78/84.6 |
| H4/C4 | 4.76/79.2 a | 4.20/70.3 | 3.77/83.8 | 3.87/82.5 | 4.22/70.6 |
| H5/C5 | 3.83/77.4 | 3.97/75.1 | 3.66/79.5 | 3.81/74.7 | 3.97/75.0 |
| H6/C6 | 3.77/63.8 | 4.24/70.1 | - | 4.41–4.33/69.0 | 4.24/69.0 |
| Bovine CS | Shark CS | |
|---|---|---|
| 4-sulfated β-GalNAc a | 68% | 16% |
| 6-sulfated β-GalNAc b | 32% | 84% |
| Dissacharides | Bovine CS | Shark CS | ||
|---|---|---|---|---|
| Capsule | Sachet | Literature Data a | Literature Data b | |
| ΔUA-GalNAc | 3 | 2 | 2 | 2 |
| ΔUA-GalNAc-6SO4 | 36 | 36 | 41 | 76 |
| ΔUA-GalNAc-4SO4 | 59 | 59 | 67 | 15 |
| ΔUA-2SO4-GalNAc-6SO4 | 1 | 1 | <1 | 7 |
| ΔUA-GalNAc-4,6diSO4 | 1 | 2 | <1 | <1 |
| ΔUA-2SO4-GalNAc-6SO4 | <1 | <1 | <1 | <1 |
| 4-sulfation of β-GalNAc | 60 | 58 | 49 | 15 |
| 6-sulfation of β-GalNAc | 38 | 39 | 35 | 83 |
| 2-sulfation of β-GlcA | 1 | 1 | <1 | 7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, G.R.C.; Piquet, A.A.; Glauser, B.F.; Tovar, A.M.F.; Pereira, M.S.; Vilanova, E.; Mourão, P.A.S. Systematic Analysis of Pharmaceutical Preparations of Chondroitin Sulfate Combined with Glucosamine. Pharmaceuticals 2017, 10, 38. https://doi.org/10.3390/ph10020038
Santos GRC, Piquet AA, Glauser BF, Tovar AMF, Pereira MS, Vilanova E, Mourão PAS. Systematic Analysis of Pharmaceutical Preparations of Chondroitin Sulfate Combined with Glucosamine. Pharmaceuticals. 2017; 10(2):38. https://doi.org/10.3390/ph10020038
Chicago/Turabian StyleSantos, Gustavo R.C., Adriana A. Piquet, Bianca F. Glauser, Ana M.F. Tovar, Mariana S. Pereira, Eduardo Vilanova, and Paulo A.S. Mourão. 2017. "Systematic Analysis of Pharmaceutical Preparations of Chondroitin Sulfate Combined with Glucosamine" Pharmaceuticals 10, no. 2: 38. https://doi.org/10.3390/ph10020038
APA StyleSantos, G. R. C., Piquet, A. A., Glauser, B. F., Tovar, A. M. F., Pereira, M. S., Vilanova, E., & Mourão, P. A. S. (2017). Systematic Analysis of Pharmaceutical Preparations of Chondroitin Sulfate Combined with Glucosamine. Pharmaceuticals, 10(2), 38. https://doi.org/10.3390/ph10020038