
Novel Proteasome Inhibitors and Histone Deacetylase Inhibitors: Progress in Myeloma Therapeutics


Abstract
:1. Introduction
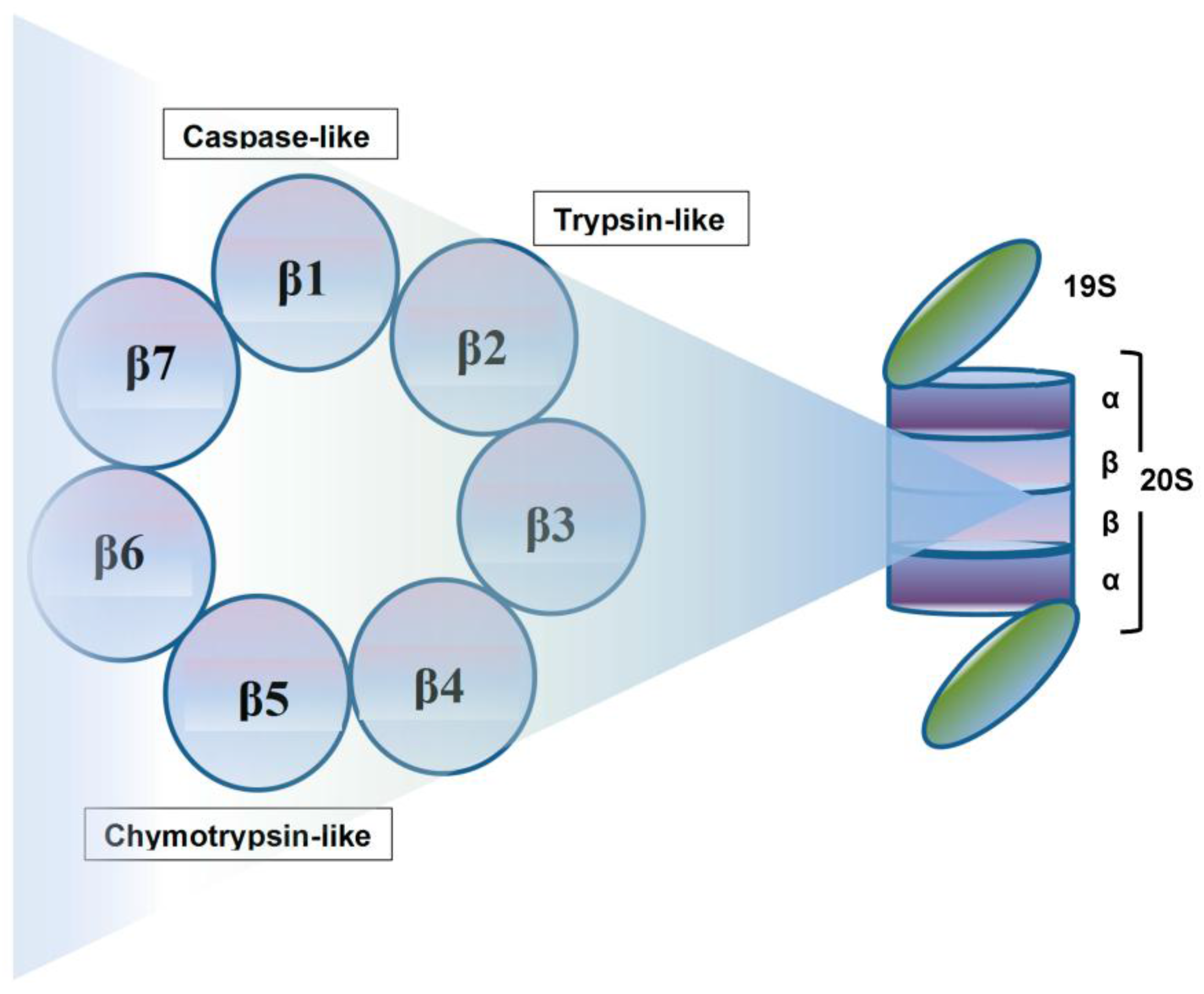
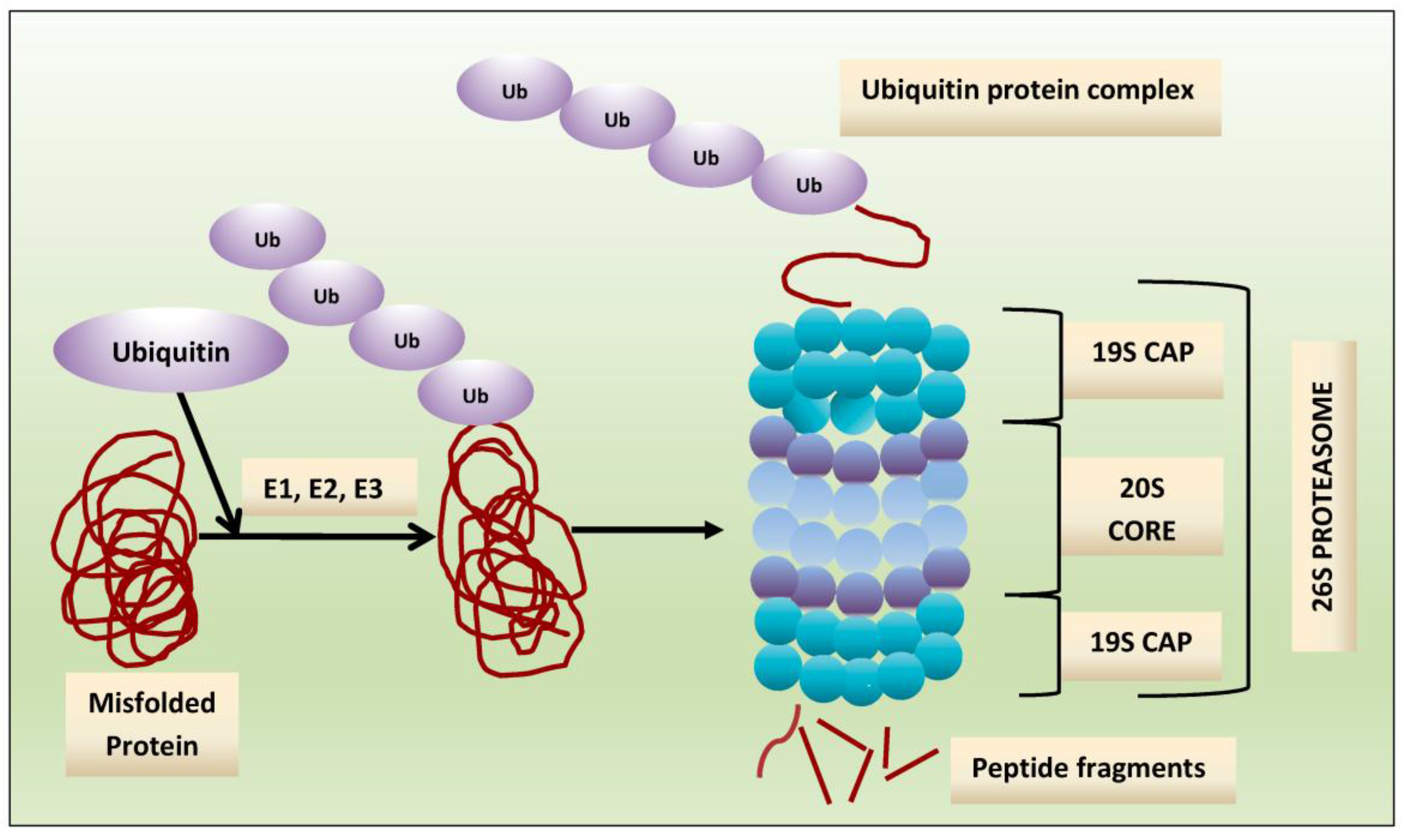
2. Proteasome: A Therapeutic Target
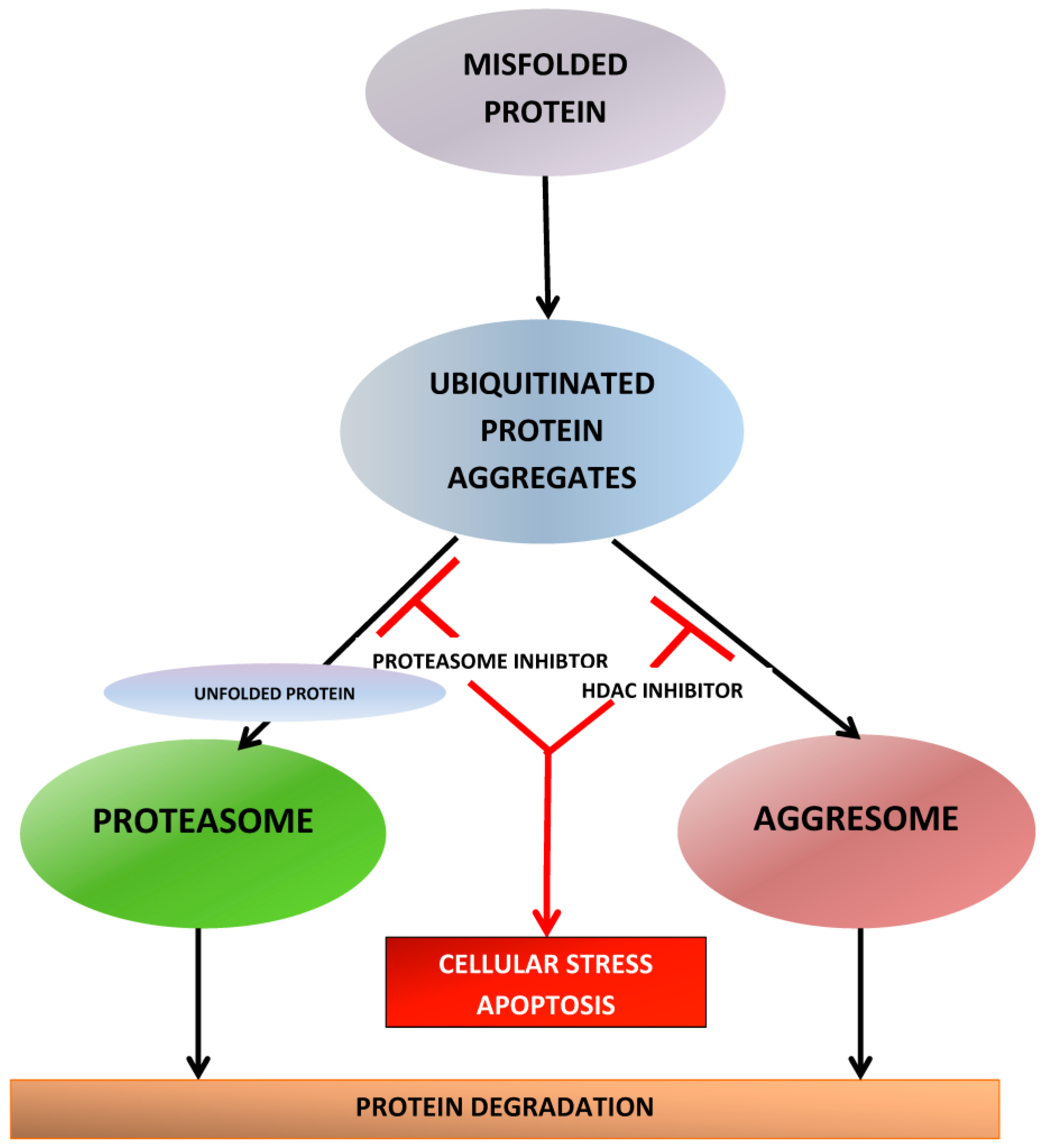
3. Proteasome Inhibitors: Mechanisms of Anti-Myeloma Effect
4. Clinical Development of Novel Proteasome Inhibitors
4.1. Carfilzomib
4.2. Ixazomib
4.3. Oprozomib
4.4. Marizomib
5. Histone Deacetylase Inhibitors
5.1. Histone Deacetylases: A Therapeutic Target
5.2. Histone Deacetylase Inhibitors: Mechanisms of Anti-Myeloma Activity
6. Clinical Development of Histone Deacetylase Inhibitors
6.1. Vorinostat
6.2. Panobinostat
6.3. Ricolinostat
7. Conclusions
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improve survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Ocio, E.M.; San Miguel, J.F. Novel generation of agents with proven clinical activity in multiple myeloma. Semin. Oncol. 2013, 40, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Hayden, P.J.; Anderson, K.C.; Richardson, P.G. From the bench to the bedside: emerging new treatments in multiple myeloma. Best Pract. Res. Clin. Haematol. 2007, 20, 797–816. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Kuhn, D.J. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 2008, 14, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Grunebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Proteasome inhibitors: antitumor effects and beyond. Leukemia 2007, 21, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Iconomou, G.; Kalofonos, H.P. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood 2008, 112, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Skrott, Z.; Cvek, B. Linking the activity of bortezomib in multiple myeloma and autoimmune diseases. Crit. Rev. Oncol. Hematol. 2014, 92, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Buac, D.; Shen, M.; Schmitt, S.; Kona, F.R.; Deshmukh, R.; Zhang, Z.; Neslund-Dudas, C.; Mitra, B.; Dou, Q.P. From bortezomib to other inhibitors of the proteasome and beyond. Curr. Pharm. Des. 2013, 19, 4025–4038. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Therapeutic advances in relapsed or refractory multiple myeloma. J. Natl. Compr. Cancer Netw. 2013, 11, 676–679. [Google Scholar] [PubMed]
- Kuhn, D.J.; Orlowski, R.Z.; Bjorklund, C.C. Second generation proteasome inhibitors: carfilzomib and immunoproteasome-specific inhibitors (IPSIs). Curr. Cancer Drug. Targets 2011, 11, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.; Gleason, C.; Casbourne, D.; Lonial, S. Relapsed and refractory lymphoid neoplasms and multiple myeloma with a focus on carfilzomib. Biologics 2013, 7, 13–32. [Google Scholar] [PubMed]
- Thompson, J.L. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann. Pharmacother. 2013, 47, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [PubMed]
- Arastu-Kapur, S.; Anderl, J.L.; Kraus, M.; Parlati, F.; Shenk, K.D.; Lee, S.J.; Muchamuel, T.; Bennett, M.K.; Driessen, C.; Ball, A.J.; et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin. Cancer Res. 2011, 17, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; San-Miguel, J.F.; Hungria, V.; Hou, J.; Moreau, P.; Lonial, S.; Lee, J.H.; Einsele, H.; Alsina, M.; Richardson, P.G. Deacetylase Inhibitors: an Advance in Myeloma Therapy? Expert Rev. Hematol. 2017, 10, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Libby, E.N.; Becker, P.S.; Burwick, N.; Green, D.J.; Holmberg, L.; Bensinger, W.I. Panobinostat: a review of trial results and future prospects in multiple myeloma. Expert Rev. Hematol. 2015, 8, 9–18. [Google Scholar] [CrossRef] [PubMed]
- El-Amm, J.; Tabbara, I.A. Emerging therapies in multiple myeloma. Am. J. Clin. Oncol. 2015, 38, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome pathway: on protein death and cell life. Embo J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). Angew. Chem. Int. Ed. Engl. 2005, 44, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Rolfe, M.; Harper, J.W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.P.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L.; Akopian, T.N.; Kisselev, A.F.; Lee, D.H.; Rohrwild, M. New insights into the mechanisms and importance of the proteasome in intracellular protein degradation. Biol. Chem. 1997, 378, 131–140. [Google Scholar] [PubMed]
- Hochstrasser, M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 1995, 7, 215–223. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca, P.C.; He, J.; Morris, E.P. Molecular model of the human 26S proteasome. Mol. Cell 2012, 46, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef]
- Jager, S.; Groll, M.; Huber, R.; Wolf, D.H.; Heinemeyer, W. Proteasome beta-type subunits: unequal roles of propeptides in core particle maturation and a hierarchy of active site function. J. Mol. Biol. 1999, 291, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Heinemeyer, W.; Fischer, M.; Krimmer, T.; Stachon, U.; Wolf, D.H. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 1997, 272, 25200–25209. [Google Scholar] [CrossRef] [PubMed]
- Lightcap, E.S.; McCormack, T.A.; Pien, C.S.; Chau, V.; Adams, J.; Elliott, P.J. Proteasome inhibition measurements: clinical application. Clin. Chem. 2000, 46, 673–683. [Google Scholar] [PubMed]
- Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Jakubowiak, A.J.; Lonial, S.; Tudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171–003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.E.; Pour, L.; Hajek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone (Kd) vs bortezomib and dexamethasone (Vd) in patients (pts) with relapsed multiple myeloma (RMM): Results from the phase III study ENDEAVOR. J. Clin. Oncol. 2015, 33, 8509. [Google Scholar]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Spicka, I.; Oriol, A.; Hajek, R.; Rosinol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R.; Cartmell, A.; Bessudo, A.; Lyons, R.M.; Harb, W.; Tzachanis, D.; Agajanian, R.; Boccia, R.; Coleman, M.; Moss, R.A.; et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016, 127, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Hajek, R.; Masszi, T.; Petrucci, M.T.; Palumbo, A.; Rosinol, L.; Nagler, A.; Yong, K.L.; Oriol, A.; Minarik, J.; Pour, L.; et al. A randomized phase III study of carfilzomib vs low-dose corticosteroids with optional cyclophosphamide in relapsed and refractory multiple myeloma (FOCUS). Leukemia 2017, 31, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Hacker, E.; Zweegman, S.; Kersten, M.J.; Vellenga, E.; van Marwijk-Kooy, M.; de Weerdt, O.; Lonergan, S.; Palumbo, A.; Lokhorst, H.M. Carfilzomib combined with thalidomide and dexamethasone (CARTHADEX) as induction treatment prior to high-dose melphalan (HDM) in newly diagnosed patients with multiple myeloma(MM). A trial of the European Myeloma Network EMN. ASH Annu. Meet. Abstr. 2011, 118, 633. [Google Scholar]
- Mikhael, J.R.; Reeder, C.B.; Libby, E.N.; Costa, L.J.; Bergsagel, P.L.; Buadi, F.; Mayo, A.; Nagi Reddy, S.K.; Gano, K.; Dueck, A.C.; et al. Phase Ib/II trial of CYKLONE (cyclophosphamide, carfilzomib, thalidomide and dexamethasone) for newly diagnosed myeloma. Br. J. Haematol. 2015, 169, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Bringhen, S.; Petrucci, M.T.; Larocca, A.; Conticello, C.; Rossi, D.; Magarotto, V.; Musto, P.; Boccadifuoco, L.; Offidani, M.; Omede, P.; et al. Carfilzomib, cyclophosphamide, and dexamethasone in patients with newly diagnosed multiple myeloma: a multicenter, phase 2 study. Blood 2014, 124, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.J.; Dytfeld, D.; Griffith, K.A.; Lebovic, D.; Vesole, D.H.; Jagannath, S.; Al-Zoubi, A.; Anderson, T.; Nordgren, B.; Detweiler-Short, K.; et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012, 120, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Dimopoulos, M.; Jagannath, S.; Goldschmidt, H.; Durrant, S.; Kaufman, J.L.; Leleu, X.; Nagler, A.; Offner, F.; Graef, T.; et al. VANTAGE 095: An International, Multicenter, Open-Label Study of Vorinostat (MK-0683) in Combination with Bortezomib in Patients With Relapsed and Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, 329–334. [Google Scholar] [PubMed]
- Dimopoulos, M.; Siegel, D.S.; Lonial, S.; Qi, J.; Hajek, R.; Facon, T.; Rosinol, L.; Williams, C.; Blacklock, H.; Goldschmidt, H.; et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 2013, 14, 1129–1140. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): a randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; York, I.A.; Saric, T.; Goldberg, A.L. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 2002, 80, 1–70. [Google Scholar] [PubMed]
- Rivett, A.J.; Hearn, A.R. Proteasome function in antigen presentation: immunoproteasome complexes, Peptide production, and interactions with viral proteins. Curr. Protein Pept. Sci. 2004, 5, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.M.; Ossendorp, F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr. Opin. Immunol. 2004, 16, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [PubMed]
- Arendt, C.S.; Hochstrasser, M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7156–7161. [Google Scholar] [CrossRef] [PubMed]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Luciani, F.; Kesmir, C.; Mishto, M.; Or-Guil, M.; de Boer, R.J. A mathematical model of protein degradation by the proteasome. Biophys. J. 2005, 88, 2422–2432. [Google Scholar] [CrossRef] [PubMed]
- Wiest, D.L.; Burkhardt, J.K.; Hester, S.; Hortsch, M.; Meyer, D.I.; Argon, Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. J. Cell. Biol. 1990, 110, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Cenci, S.; Mezghrani, A.; Cascio, P.; Bianchi, G.; Cerruti, F.; Fra, A.; Lelouard, H.; Masciarelli, S.; Mattioli, L.; Oliva, L.; et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. Embo J. 2006, 25, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell. Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl Acad. Sci USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.W.; Hendershot, L.M.; Sherr, C.J.; Diehl, J.A. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc. Natl Acad. Sci. USA 1999, 96, 8505–8510. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Herrmann, M.; et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Molineaux, S.M. Molecular pathways: targeting proteasomal protein degradation in cancer. Clin. Cancer Res. 2012, 18, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Franken, B.; van de Donk, N.W.; Cloos, J.C.; Zweegman, S.; Lokhorst, H.M. A clinical update on the role of carfilzomib in the treatment of relapsed or refractory multiple myeloma. Ther. Adv. Hematol. 2016, 7, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, S.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Raasi, S.; Wolf, D.H. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 2007, 18, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Baldwin, A.S. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002, 8, 385–389. [Google Scholar] [CrossRef]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.; Libermann, T.A.; Anderson, K.C. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood 1996, 87, 1104–1112. [Google Scholar] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Richardson, P.G.; Hideshima, T.; Munshi, N.; Treon, S.P.; Anderson, K.C. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood 2002, 99, 4079–4086. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; Poulaki, V.; Tai, Y.T.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood 2003, 101, 2377–2380. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. Proteasome inhibition as a novel therapeutic target in human cancer. J. Clin. Oncol. 2005, 23, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.R.; Ahmed, M.; Ahmed, S.O.; Al-Thari, S.; Khan, A.S.; Razack, S.; Platanias, L.C.; Al-Kuraya, K.S.; Uddin, S. Proteasome inhibitor MG-132 mediated expression of p27Kip1 via S-phase kinase protein 2 degradation induces cell cycle coupled apoptosis in primary effusion lymphoma cells. Leuk. Lymphoma 2009, 50, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.J.; Walker, B.; Irvine, A.E. Proteasome inhibitors in cancer therapy. J. Cell. Commun. Signal. 2011, 5, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, O.A.; Stewart, A.K.; Vallone, M.; Molineaux, C.J.; Kunkel, L.A.; Gerecitano, J.F.; Orlowski, R.Z. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res. 2009, 15, 7085–7091. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Martin, T.; Nooka, A.; Harvey, R.D.; Vij, R.; Niesvizky, R.; Badros, A.Z.; Jagannath, S.; McCulloch, L.; Rajangam, K.; et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase II clinical studies. Haematologica 2013, 98, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Mateos, M.V.; San-Miguel, J.F. Novel agents derived from the currently approved treatments for MM: novel proteasome inhibitors and novel IMIDs. Expert Opin Investig. Drugs 2012, 21, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, S.; Vij, R.; Stewart, A.K.; Trudel, S.; Jakubowiak, A.J.; Reiman, T.; Somlo, G.; Bahlis, N.; Lonial, S.; Kunkel, L.A.; et al. An open-label single-arm pilot phase II study (PX-171–003-A0) of low-dose, single-agent carfilzomib in patients with relapsed and refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2012, 12, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Lee, J.H.; Lahuerta, J.J.; Morgan, G.; Richardson, P.G.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 2012, 26, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Wang, M.; Kaufman, J.L.; Lonial, S.; Jakubowiak, A.J.; Stewart, A.K.; Kukreti, V.; Jagannath, S.; McDonagh, K.T.; Alsina, M.; et al. An open-label, single-arm, phase 2 (PX-171–004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 2012, 119, 5661–5670. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Siegel, D.S.; Vesole, D.H.; Lee, P.; Rosen, S.T.; Zojwalla, N.; Holahan, J.R.; Lee, S.; Wang, Z.; Badros, A. Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. J. Clin. Oncol. 2015, 33, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Lendvai, N.; Hilden, P.; Devlin, S.; Landau, H.; Hassoun, H.; Lesokhin, A.M.; Tsakos, I.; Redling, K.; Koehne, G.; Chung, D.J.; et al. A phase 2 single-center study of carfilzomib 56 mg/m2 with or without low-dose dexamethasone in relapsed multiple myeloma. Blood 2014, 124, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Siegel, S.D.; Martin, T.; Vij, R.; Wang, L.; Jakubowiak, A.J.; Lonial, S.; Kukreti, V.; Zonder, J.A.; Wong, A.F.; et al. Integrated safety from phase 2 studies of monotherapy carfilzomib in patients with relapsed and refractory multiple myeloma (MM): an updated analysis. ASH Annu. Meet. Abstr. 2011, 118, 1876. [Google Scholar]
- Badros, A.Z.; Vij, R.; Martin, T.; Zonder, J.A.; Kunkel, L.; Wang, Z.; Lee, S.; Wong, A.F.; Niesvizky, R. Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia 2013, 27, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.L. Treatment of relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 1774. [Google Scholar] [PubMed]
- Muchtar, E.; Gertz, M.A.; Magen, H. A practical review on carfilzomib in multiple myeloma. Eur. J. Haematol. 2016, 96, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.J.; Stadtmauer, E.A.; Abonour, R.; Cohen, A.D.; Bensinger, W.I.; Gasparetto, C.; Kaufman, J.L.; Lentzsch, S.; Vogl, D.T.; Gomes, C.L.; et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015, 126, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Korde, N.; Roschewski, M.; Zingone, A.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Tageja, N.; Kazandjian, D.; Mailankody, S.; Wu, P.; et al. Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA Oncol. 2015, 1, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Asselbergs, E.; Zweegman, S.; van der Holt, B.; Kersten, M.J.; Vellenga, E.; van Marwijk-Kooy, M.; Broyl, A.; de Weerdt, O.; Lonergan, S.; et al. Phase 2 study of carfilzomib, thalidomide, and dexamethasone as induction/consolidation therapy for newly diagnosed multiple myeloma. Blood 2015, 125, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Orlowski, R.Z. The immunoproteasome as a target in hematologic malignancies. Semin. Hematol. 2012, 49, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; Ghazarian, R.N.; Ou, M.; Luderer, M.J.; Kusdono, H.D.; Azab, A.K. Spotlight on ixazomib: potential in the treatment of multiple myeloma. Drug Des. Devel. Ther. 2016, 10, 217–226. [Google Scholar] [PubMed]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Zhan, Q.; Bae, I.; Fornace, A.J., Jr. Mammalian GADD34, an apoptosis- and DNA damage-inducible gene. J. Biol. Chem. 1997, 272, 13731–13737. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhao, J.J.; Tai, Y.T.; Amin, S.B.; Hu, Y.; Berger, A.J.; Richardson, P.; Chauhan, D.; Anderson, K.C. Investigational agent MLN9708/2238 targets tumor-suppressor miR33b in MM cells. Blood 2012, 120, 3958–3967. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Offidani, M.; Vigna, E.; Corvatta, L.; Recchia, A.G.; Morabito, L.; Morabito, F.; Gentili, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2015, 24, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Guohui, G.; Berg, D.; Kalebic, T.; Gomez-Navarro, J. Clinical pharmacokinetics of intravenous and oral MLN9708, an investigational proteasome inhibitor: an analysis of data from four phase 1 monotherapy studies. Blood 2010, 116, 1813. [Google Scholar]
- Allegra, A.; Alonci, A.; Gerace, D.; Russo, S.; Innao, V.; Calabro, L.; Musolino, C. New orally active proteasome inhibitors in multiple myeloma. Leuk. Res. 2014, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.E.; Chang, J.; Cheson, B.D.; Rifkin, R.; Hamburg, S.; Reyes, R.; Hui, A.M.; Yu, J.; Gupta, N.; Di Bacco, A.; et al. Phase 1 dose-escalation study of IV ixazomib, an investigational proteasome inhibitor, in patients with relapsed/refractory lymphoma. Blood Cancer J. 2014, 4, e251. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Bensinger, W.I.; Zimmerman, T.M.; Reeder, C.B.; Berenson, J.R.; Berg, D.; Hui, A.M.; Gupta, N.; Di Bacco, A.; Yu, J.; et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood 2014, 124, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Baz, R.; Wang, M.; Jakubowiak, A.J.; Laubach, J.P.; Harvey, R.D.; Talpaz, M.; Berg, D.; Liu, G.; Yu, J.; et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood 2014, 124, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Roy, V.; Reeder, C.B.; Lacy, M.Q.; Gertz, M.A.; Laumann, K.; Thompson, M.A.; Witzig, T.E.; Buadi, F.K.; et al. Phase 2 trial of ixazomib in patients with relapsed multiple myeloma not refractory to bortezomib. Blood Cancer J. 2015, 4, e338. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Zhao, Y.; Hui, A.M.; Esseltine, D.L.; Venkatakrishnan, K. Switching from body surface area-based to fixed dosing for the investigational proteasome inhibitor ixazomib: a population pharmacokinetic analysis. Br. J. Clin. Pharmacol. 2015, 79, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Handa, H.; Suzuki, K.; Chou, T.; Matsushima, T. Phase 1 study of the investigational proteasome inhibitor ixazomib alone or in combination with lenalidomide-dexamethasone in Japanese patients with relapsed and/or refractiory multiple myeloma. Blood 2014, 124, 5752. [Google Scholar]
- Richardson, P.G.; Weller, E.; Lonial, S.; Jakubowiak, A.J.; Jagannath, S.; Raje, N.S.; Avigan, D.E.; Xie, W.; Ghobrial, I.M.; Schlossman, R.L.; et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 2010, 116, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Berdeja, J.G.; Niesvizky, R.; Lonial, S.; Laubach, J.P.; Hamadani, M.; Stewart, A.K.; Hari, P.; Roy, V.; Vescio, R.; et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014, 15, 1503–1512. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hofmeister, C.C.; Rosenbaum, C.A.; Htut, M.; Vesole, D.H.; Berdeja, J.; Liedtke, M.; Chari, A.; Smith, S.D.; Lebovic, D.; et al. Twice-weekly oral MLN9708 (ixazomib citrate), an investigational proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma: final phase 1 results and phase 2 data. Blood 2013, 122, 535. [Google Scholar]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: a review about the future. J. Hematol. Oncol. 2016, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Grosicki, S.; Jedrzejczak, W.W.; Nahi, H.; Gruber, A.; Hansson, M.; Byrne, C.; Labotka, R.; Hui, A.-M.; Teng, Z.; et al. Randomized Phase 2 Study of the All-Oral Combination of Investigational Proteasome Inhibitor (PI) Ixazomib Plus Cyclophosphamide and Low-Dose Dexamethasone (ICd) in Patients (Pts) with Newly Diagnosed Multiple Myeloma (NDMM) Who Are Transplant-Ineligible (NCT02046070). Blood 2015, 126, 26. [Google Scholar]
- Kumar, S.; Berdeja, J.; Niesvizky, R.; Lonial, S.; Laubach, J.P.; Hamadani, M.; Stewart, A.K.; Hari, P.N.; Roy, R.; Vescio, R.; et al. Long-Term Ixazomib Maintenance Is Tolerable and Improves Depth of Response Following Ixazomib-Lenalidomide-Dexamethasone Induction in Patients (Pts) with Previously Untreated Multiple Myeloma (MM): Phase 2 Study Results. Blood 2014, 124, 82. [Google Scholar]
- Attal, M.; Lauwers-Cances, V.; Marit, G.; Caillot, D.; Moreau, P.; Facon, T.; Stoppa, A.M.; Hulin, C.; Benboubker, L.; Garderet, L.; et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 2012, 366, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, P.L.; Owzar, K.; Hofmeister, C.C.; Hurd, D.D.; Hassoun, H.; Richardson, P.G.; Giralt, S.; Stadtmauer, E.A.; Weisdorf, D.J.; Vij, R.; et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 2012, 366, 1770–1781. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.J.; Baladandayuthapani, V.; Weber, D.M.; Thomas, S.K.; Alexanian, R.; Wang, M.; Qazilbash, M.H.; Champlin, R.E.; Shah, N.; Bashir, Q.; et al. Phase II study of the combination of MLN9708 with lenalidomide as maintenance therapy post autologous stem cell transplant in patients with multiple myeloma. Blood 2013, 122, 1983. [Google Scholar]
- Zhou, H.J.; Aujay, M.A.; Bennett, M.K.; Dajee, M.; Demo, S.D.; Fang, Y.; Ho, M.N.; Jiang, J.; Kirk, C.J.; Laidig, G.J.; et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.M.; Kumar, S. New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. 2016, 6, e451. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Aujay, M.; Ngo, H.T.; Azab, A.K.; Azab, F.; Quang, P.; Maiso, P.; Runnels, J.; Anderson, K.C.; et al. Selective inhibition of chymotrypsin-like activity of the immunoproteasome and constitutive proteasome in Waldenstrom macroglobulinemia. Blood 2010, 115, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. The next generation proteasome inhibitors carfilzomib and oprozomib activate prosurvival autophagy via induction of the unfolded protein response and ATF4. Autophagy 2012, 8, 1873–1874. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Savona, M.; Siegel, D.S.; Kaufman, J.L.; Badros, A.; Ghobrial, I.M.; Paner, A.; Jagannath, S.; Jakubowiak, A.; Mikhael, J.R.; et al. Clinical profile of single-agent oprozomib in patients (Pts) with multiple myeloma (MM): updated results from a multicenter, open-label, dose escalation phase 1b/2 study. Blood 2014, 124, 34. [Google Scholar]
- Hari, P.N.; Shain, K.; Voorhees, P.M.; Gabrail, N.; Abidi, M.H.; Zonder, J.; Boccia, R.V.; Richardson, P.G.; Neuman, L.L.; Dixon, S.J.; et al. Oprozomib and dexamethasone in patients with relapsed and/or refractory multiple myeloma: initial results from the dose escalation portion of a phase 1b/2, Multicenter, Open-Label Study. Blood 2014, 124, 3453. [Google Scholar]
- Shah, J.; Niesvizky, R.; Stadtmauer, E.; Rifkin, R.M.; Berenson, J.; Berdeja, J.; Sharman, J.P.; Lyons, R.; Klippel, Z.; Wong, H.; et al. Oprozomib, pomalidomide, and dexamethasone (OPomd) in patients (Pts) with relapsed and/or refractory multiple myeloma (RRMM): initial results of a phase 1b study. Blood 2015, 126, 378. [Google Scholar]
- A Study of Oprozomib, Pomalidomide, and Dexamethasone in Subjects With Primary Refractory or Relapsed and Refractory Multiple Myeloma. Available online: clinicaltrials.gov/ct2/show/NCT01999335 (Accessed on 10 April 2017).
- Fenical, W.; Jensen, P.R.; Palladino, M.A.; Lam, K.S.; Lloyd, G.K.; Potts, B.C. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg. Med. Chem. 2009, 17, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Hideshima, T.; Anderson, K.C. A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Br. J. Cancer 2006, 95, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew. Chem. Int. Ed. Engl. 2003, 42, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Lawasut, P.; Chauhan, D.; Laubach, J.; Hayes, C.; Fabre, C.; Maglio, M.; Mitsiades, C.; Hideshima, T.; Anderson, K.C.; Richardson, P.G. New proteasome inhibitors in myeloma. Curr. Hematol. Malig. Rep. 2012, 7, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.; Brahmandam, M.; Podar, K.; Hideshima, T.; Richardson, P.; Munshi, N.; Palladino, M.A.; Anderson, K.C. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 2008, 111, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.L.; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004, 10, 3954–3964. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Suzuki, E.; Umezawa, K.; Spandidos, D.A.; Berenson, J.; Daniels, T.R.; Penichet, M.L.; Jazirehi, A.R.; Palladino, M.; Bonavida, B. Inhibition of Yin Yang 1-dependent repressor activity of DR5 transcription and expression by the novel proteasome inhibitor NPI-0052 contributes to its TRAIL-enhanced apoptosis in cancer cells. J. Immunol. 2008, 180, 6199–6210. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Ciccarelli, B.; Richardson, P.G.; Palladino, M.A.; Anderson, K.C. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood 2010, 115, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Zimmerman, T.M.; Hofmeister, C.C.; Talpaz, M.; Chanan-Khan, A.A.; Kaufman, J.L.; Laubach, J.P.; Chauhan, D.; Jakubowiak, A.J.; Reich, S.; et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052–101 Part 1. Blood 2016, 127, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase I Clinical Trial of Marizomib (NPI-0052) in Patients with Advanced Malignancies Including Multiple Myeloma: Study NPI-0052–102 Final Results. Clin. Cancer Res. 2016, 22, 4559–4566. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Harrison, S.; Laubach, J.; Zonder, J.; Badros, A.; Bergin, K.; Khot, A.; Zimmerman, T.; Anderson, K.C.; MacLaren, A.; et al. Pmd-107: Marizomib, Pomalidomide and Low Dose-Dexamethasone Combination Study in Relapsed/Refractory Multiple Myeloma (NCT02103335): Full Enrollment Results from a Phase-1 Multicenter, Open Label Study. Blood 2016, 128, 3326. [Google Scholar]
- Badros, A.Z.; Singh, Z.; Dhakal, B.; Kwok, Y.; MacLaren, A.; Richardson, P.G.; Trikha, M.; Parameswaran, H. Marizomib for CNS-Multiple Myeloma. Blood 2016, 128, 2118. [Google Scholar]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. Epigenetic therapies reach main street. Clin. Cancer Res. 2009, 15, 3917. [Google Scholar] [CrossRef] [PubMed]
- Batty, N.; Malouf, G.G.; Issa, J.P. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009, 280, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Zain, J. Role of histone deacetylase inhibitors in the treatment of lymphomas and multiple myeloma. Hematol. Oncol. Clin. N. Am. 2012, 26, 671–704. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Stimson, L.; Wood, V.; Khan, O.; Fotheringham, S.; La Thangue, N.B. HDAC inhibitor-based therapies and haematological malignancy. Ann. Oncol. 2009, 20, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Fabre, C.; Lonial, S.; Richardson, P.G. Histone deacetylase inhibitors in multiple myeloma: rationale and evidence for their use in combination therapy. Clin. Lymphoma Myeloma Leuk. 2013, 13, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Moreau, P.; Laubach, J.P.; Maglio, M.E.; Lonial, S.; San-Miguel, J. Deacetylase inhibitors as a novel modality in the treatment of multiple myeloma. Pharmacol. Res. 2016, 117, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology 2011, 31, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Panobinostat for the treatment of multiple myeloma. Lancet Oncol. 2014, 15, 1178–1179. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Jones, S.S.; Spencer, A. Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br. J. Haematol. 2013, 162, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Xu, W.; Ngo, L.; Perez, G.; Dokmanovic, M.; Marks, P.A. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc. Natl. Acad. Sci. USA 2006, 103, 15540–15545. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Gobbi, M.; Patrone, F.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. New insights into the treatment of multiple myeloma with histone deacetylase inhibitors. Curr. Pharm. Des. 2013, 19, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29, 3–9. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Hideshima, T.; Anderson, K.C. Histone deacetylase inhibitors in multiple myeloma: from bench to bedside. Int. J. Hematol. 2016, 104, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Wada, T.; Shimizu, R.; Izumi, T.; Akutsu, M.; Mitsunaga, K.; Noborio-Hatano, K.; Nobuyoshi, M.; Ozawa, K.; Kano, Y.; et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 2010, 116, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Hideshima, T.; Akiyama, M.; Chauhan, D.; Munshi, N.; Gu, X.; et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl Acad. Sci. USA 2004, 101, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Catley, L.; Weisberg, E.; Tai, Y.T.; Atadja, P.; Remiszewski, S.; Hideshima, T.; Mitsiades, N.; Shringarpure, R.; LeBlanc, R.; Chauhan, D.; et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 2003, 102, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.B.; Maududi, T.; Barton, K.; Ayers, J.; Alkan, S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br. J. Haematol. 2004, 125, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; McMullan, C.; Poulaki, V.; Fanourakis, G.; Schlossman, R.; Chauhan, D.; Munshi, N.C.; Hideshima, T.; et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood 2003, 101, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Mitsiades, C.; Colson, K.; Reilly, E.; McBride, L.; Chiao, J.; Sun, L.; Ricker, J.; Rizvi, S.; Oerth, C.; et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk. Lymphoma 2008, 49, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K. Novel therapeutics in multiple myeloma. Hematology 2012, 17, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Vesole, D.H.; Jagannath, S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: a case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010, 10, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, S.; Dimopoulos, M.A.; Lonial, S. Combined proteasome and histone deacetylase inhibition: A promising synergy for patients with relapsed/refractory multiple myeloma. Leuk. Res. 2010, 34, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.M.; Graef, T.; Hussein, M.; Sobecks, R.M.; Schiller, G.J.; Lupinacci, L.; Hardwick, J.S.; Jagannath, S. Phase I trial of vorinostat combined with bortezomib for the treatment of relapsing and/or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2012, 12, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Richardson, P.; Dimopoulos, M.; Moreau, P.; Mitsiades, C.; Weber, D.; Houp, J.; Gause, C.; Vuocolo, S.; Eid, J.; et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J. 2014, 4, e182. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.; Vesole, D.H.; Richter, J.R.; Biran, N.; Bilotti, E.; McBride, L.; Anand, P.; Ivanovski, K.; Siegel, D.S. A phase IIb trial of vorinostat in combination with lenalidomide and dexamethasone in patients with multiple myeloma refractory to previous lenalidomide-containing regimens. Br. J. Haematol. 2017, 176, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Vesole, D.H.; Bilotti, E.; Richter, J.R.; McNeill, A.; McBride, L.; Raucci, L.; Anand, P.; Bednarz, U.; Ivanovski, K.; Smith, J.; et al. Phase I study of carfilzomib, lenalidomide, vorinostat, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Br. J. Haematol. 2015, 171, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Pan, Y.; Smyth, G.K.; George, D.J.; McCormack, C.; Williams-Truax, R.; Mita, M.; Beck, J.; Burris, H.; Ryan, G.; et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin. Cancer Res. 2008, 14, 4500–4510. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Spencer, A.; Bhalla, K.N.; Prince, H.M.; Fischer, T.; Kindler, T.; Giles, F.J.; Scott, J.W.; Parker, K.; Liu, A.; et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia 2013, 27, 1628–1636. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.L.; Siegel, D.; Goldschmidt, H.; Hazell, K.; Bourquelot, P.M.; Bengoudifa, B.R.; Matous, J.; Vij, R.; de Magalhaes-Silverman, M.; Abonour, R.; et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk. Lymphoma 2012, 53, 1820–1823. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Vilanova, D.; Atadja, P.; Maiso, P.; Crusoe, E.; Fernandez-Lazaro, D.; Garayoa, M.; San-Segundo, L.; Hernandez-Iglesias, T.; de Alava, E.; et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010, 95, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Richardson, P.G.; Gunther, A.; Sezer, O.; Siegel, D.; Blade, J.; LeBlanc, R.; Sutherland, H.; Sopala, M.; Mishra, K.K.; et al. Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J. Clin. Oncol. 2013, 31, 3696–3703. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Panobinostat: first global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Guenther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: outcomes by prior treatment. Blood 2016, 127, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Hart, L.L.; Mace, J.R.; Arrowsmith, E.R.; Essell, J.H.; Owera, R.S.; Hainsworth, J.D.; Flinn, I.W. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015, 100, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Redic, K.A.; Hough, S.M.; Price, E.M. Clinical developments in the treatment of relapsed or relapsed and refractory multiple myeloma: impact of panobinostat, the first-in-class histone deacetylase inhibitor. OncoTargets Ther. 2016, 9, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.J.; Thomas, S.K.; Weber, D.M.; Wang, M.; Alexanian, R.; Qazilbash, M.H.; Bashir, Q.; Parmar, S.; Shah, N.; Popat, U.R.; et al. Phase 1/1b Study of the Efficacy and Safety of the Combination of Panobinostat + Carfilzomib in Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 2012, 120, 4081. [Google Scholar]
- Kaufman, J.; Zimmerman, T.; Rosenbaus, C.; Nooka, A.K.; Heffner, L.T.; Harvey, R.D.; Gleason, C.; Lewis, C.; Sharp, C.; Barron, K.W.; et al. Phase I study of the combination of carfilzomib and panobinostat for patients with relapsed and refractory myeloma: a Multiple Myeloma Research Consortium (MMRC) clinical trial. Blood 2014, 124, 32. [Google Scholar]
- Reu, F.J.; Valent, J.; Malek, E.; Sobecks, R.M.; Faiman, B.M.; Hamilton, K.; Elberson, J.; Fada, S.; Liu, H.K.; Samaras, C.; et al. A Phase I Study of Ixazomib in Combination with Panobinostat and Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2015, 126, 4221. [Google Scholar]
- Hideshima, T.; Cottini, F.; Ohguchi, H.; Jakubikova, J.; Gorgun, G.; Mimura, N.; Tai, Y.T.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015, 5, e312. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Cho, H.J.; Leng, S.; Dhadwal, A.; Morgan, G.; La, L.; Garcia, K.; Carter, C.; Catamero, D.; Escalon, J.; et al. A phase II study of Panobinostat with lenalidomide and weekly dexamethasone in myeloma. Blood 2015, 126, 4226. [Google Scholar]
- Laubach, J.; Tuchman, S.A.; Rosenblatt, J.; Redd, R.; Colson, K.; Motta, A.; Fitzpatrick, K.; Weller, E.; Richardson, P.G. Phase 1b study of panobinostat in combination with lenalidomide, bortezomib, and dexamethasone in relapsed refractory multiple myeloma. ASCO Meet. Abstr. 2016, 34, 8014. [Google Scholar]
- Shah, J.J.; Feng, L.; Manasanch, E.E.; Weber, D.; Thomas, S.K.; Turturro, F.; Shah, N.; Popat, U.R.; Nieto, Y.; Bashir, Q.; et al. Phase I/II trial of the efficacy and safety of combination therapy with lenalidomide/bortezomib/dexamethasone (RVD) and panobinostat in transplant-eligible patients with newly diagnosed multiple myeloma. Blood 2015, 126, 187. [Google Scholar]
- Popat, R.; Brown, S.R.; Flanagan, L.; Hall, A.; Gregory, W.; Kishore, B.; Streetly, M.; Oakervee, H.; Yong, K.; Cook, G.; et al. Bortezomib, thalidomide, dexamethasone, and panobinostat for patients with relapsed multiple myeloma (MUK-six): a multicentre, open-label, phase 1/2 trial. Lancet Haematol. 2016, 3, e572–e580. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.S.; Bensinger, W.; Cole, C.; Lonial, S.; Jagannath, S.; Arce-Lara, C.E.; Valent, J.; Rosko, A.E.; Harb, W.A.; Sandhu, I.; et al. Ricolinostat (ACY-1215), the First Selective HDAC6 Inhibitor, Combines Safely with Pomalidomide and Dexamethasone and Shows Promosing Early Results in Relapsed-and-Refractory Myeloma (ACE-MM-102 Study). Blood 2015, 126, 4228. [Google Scholar]
- Vogl, D.T.; Raje, N.S.; Jagannath, S.; Richardson, P.G.; Hari, P.; Orlowski, R.Z.; Supko, J.; Tamang, D.; Jones, S.S.; Wheeler, C.; et al. Ricolinostat (ACY-1215), the first selective HDAC6 inhibitor, in combination with bortezomib and dexamethasone in patients with relapsed or relapsed-and-refractory multiple myeloma: phase 1b results (ACY-100 Study). Blood 2015, 126, 1827. [Google Scholar]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Afifi, S.; Michael, A.; Azimi, M.; Rodriguez, M.; Lendvai, N.; Landgren, O. Role of Histone Deacetylase Inhibitors in Relapsed Refractory Multiple Myeloma: A Focus on Vorinostat and Panobinostat. Pharmacotherapy 2015, 35, 1173–1188. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carfilzomib | Ixazomib | Oprozomib | Marizomib | |
|---|---|---|---|---|
| Active moiety | Epoxyketone | Boronate | Epoxyketone | Beta-lactone |
| Proteasome binding | Irreversible | Reversible | Irreversible | Irreversible |
| Proteasome inhibition | Chymotrypsin-like >> caspase-and trypsin-like | Chymotrypsin-like > caspase- and trypsin-like | Chymotrypsin-like >> caspase-and trypsin-like | Chymotrypsin- and trypsin-like >> caspase-like |
| Plasma half-life | <30 min | 18 min | <30 min | <5 min |
| Mode of administration | IV | Oral | Oral | IV |
| Status | FDA-approved with LEN-DEX as 2nd line and single-agent as 3rd line | FDA approved with LEN-DEX as 2nd line | Phase I | Phase I |
| Unique off-target organ toxicity | Cardiovascular | GI, myelosuppression, neurotoxicity | GI, myelosuppression | Central nervous system |
| Investigator [Ref.] Study | Phase | Disease Status | #Prior Therapies Allowed | Treatment | Dosing Schedule of PI | N | Survival End-Point (Months or %) | ORR | ≥VGPR Rate | Median Number of Prior Therapies | Prior Exposure | AEs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Siegel DS [36] PX-171-003-A1 | II | RRMM | ≥2 | K (single-agent) | 20/27 mg/m2 days 1, 2, 8, 9, 15, 16 q28days | 266 | mPFS 3.7, mOS 15.6 | 23.7% | 5.5% | 5 | Prior BTZ 99.6% (73% refractory), prior AHCT 74% | NR |
| Dimopoulos MA [37] ENDEAVOR | III | RRMM | 1–3 | Kd vs. Vd | 20/56 mg/m2 days 1, 2, 8, 9, 15, 16 q28days | 929 | mPFS 18.7 vs. 9.4 | 77% vs. 63% | 54% vs. 29% | 2 | Previous BTZ 54% vs. 54% | SAE: 48% vs. 36% |
| Stewart AK [38] ASPIRE | III | RRMM | 1–3 | KRd vs. Rd | 20/27 mg/m2 days 1, 2, 8, 9, 15, 16 q28days | 792 | mPFS 26.3 vs. 17.6 | 87% vs. 67% | 70% vs. 40% | 2 | Previous BTZ 66% vs. 66% | SAE: 60% vs. 54% |
| Berenson JR [39] CHAMPION | I/II | RRMM | 1–3 | Kd | MTD 70 mg/m2 days 1, 8, 15 q28days | 116 | mPFS 12.6 | 77% | 33% | 1 | BTZ-refract 55% | SAE: 35% |
| Hajek R [40] FOCUS | III | RRMM | ≥3 | K vs. d (+Cy) | 20/27 mg/m2 days 1, 2, 8, 9, 15, 16 q28days | 315 | mPFS 3.7 vs. 3.3, mOS 10.2 vs. 10 | 19% vs. 11% | 5 | SAE: 59% vs. 51% | ||
| Sonneveld P [41] | II | NDMM | - | KTd | 20/27–56 mg/m2 days 1, 2, 8,9, 15, 16 q28days | 91 | 3-year PFS 72% | 90% | 68% after 4 cycles 89% after consolidat-ion | - | - | SAE: 40% |
| Mikhael JR [42] CYKLONE | I/II | NDMM | - | KCyTd | MTD of 20/36 mg/m2 IV d1,2,8,9,15,16 q28d | 64 | 2-year PFS 76% and OS 96% | 91% | 69% | G ≥ 3 AE: 67% | ||
| CLARION (unpublished) | III | NDMM, AHCT-ineligible | - | KMP vs. VMP | CFZ 20/36 mg/m2 IV days 1, 2, 8, 9, 22, 23, 29, 30 q42days | 955 | mPFS 22.3 vs. 22.1 | NR | NR | - | - | G ≥ 3 AEs: 75% vs. 76% |
| Bringhen S [43] | II | NDMM, AHCT-ineligible | - | KCyd | 20/36 mg/m2 IV d1, 2, 8, 9, 15, 16 q28days | 58 | 2-year PFS 76%, OS 87% | 95% | 71% | - | - | SAE: 28% |
| Jakubowiak AJ [44] | I/II | NDMM, AHCT-eligible and ineligible | - | KRd | 20/20–36 mg/m2 IV d1, 2, 8, 9, 15, 16 q28days | 53 | 3-year PFS 79.6%, OS 100% | 100% | 91% | - | - | NR |
| Moreau P [45] TOURMALINE-MM1 | III | RRMM | 1–3 | IRd vs. Placebo-Rd | 4 mg PO d1, 8, 15 q28d | 722 | mPFS 20.6 vs. 14.7 | 78% vs. 72% | 48% vs. 39% | 1; Prior auto 59% vs. 55% | Prior BTZ 69% vs. 69% Prior IMId 54% vs. 56% | SAE: 47% vs. 49% |
| Investigator [Ref.] Study | Phase | Disease Status | #Prior Therapies Allowed | Treatment | Dosing Schedule of the HDACi | N | Survival End-Point (Months or %) | ORR | ≥VGPR Rate | Median Number of Prior Therapies | Prior Exposure | AEs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Siegel DS [46] VANTAGE-095 | IIb | RRMM | ≥2 | Vorinostat-BTZ | Vorinostat 400 mg PO days 1–14 q21days | 143 | PFS 3.1, OS 11.2 | 11.3% | 1% VGPR | 2 | BTZ-refractory 100%, IMiD-refractory 87% | SAE: 65% |
| Dimopoulos M [47] VANTAGE-088 | III | RRMM | 1–3 | Vorinostat-Vd vs. placebo-Vd | Vorinostat 400 mg PO days 1–14 q21days | 637 | mPFS 7.6 vs. 6.8, mOS NA vs. 28 | 56% vs. 40% | CR 7.9% vs. 5.3% | 2 | Previous BTZ 25% vs. 23% | SAE: 41.3% vs. 43.1% |
| San Miguel JF [48,49] PANORAMA 1 | III | RRMM | 1–3 | PanoVd vs. Placebo-Vd | Pano 20 mg PO days 1, 3, 5, 8, 10, 12 q21days | 768 | mPFS 12.1 vs. 8.1, mOS 40.3 vs. 35.8 | 61% vs. 55% | 28% vs. 16% | 2 | Previous BTZ 51% vs. 52% Previous AHCT 56% vs. 59% | SAEs 60% vs. 42% |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhabra, S. Novel Proteasome Inhibitors and Histone Deacetylase Inhibitors: Progress in Myeloma Therapeutics. Pharmaceuticals 2017, 10, 40. https://doi.org/10.3390/ph10020040
Chhabra S. Novel Proteasome Inhibitors and Histone Deacetylase Inhibitors: Progress in Myeloma Therapeutics. Pharmaceuticals. 2017; 10(2):40. https://doi.org/10.3390/ph10020040
Chicago/Turabian StyleChhabra, Saurabh. 2017. "Novel Proteasome Inhibitors and Histone Deacetylase Inhibitors: Progress in Myeloma Therapeutics" Pharmaceuticals 10, no. 2: 40. https://doi.org/10.3390/ph10020040
APA StyleChhabra, S. (2017). Novel Proteasome Inhibitors and Histone Deacetylase Inhibitors: Progress in Myeloma Therapeutics. Pharmaceuticals, 10(2), 40. https://doi.org/10.3390/ph10020040