
Exploring Wound-Healing Genomic Machinery with a Network-Based Approach

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
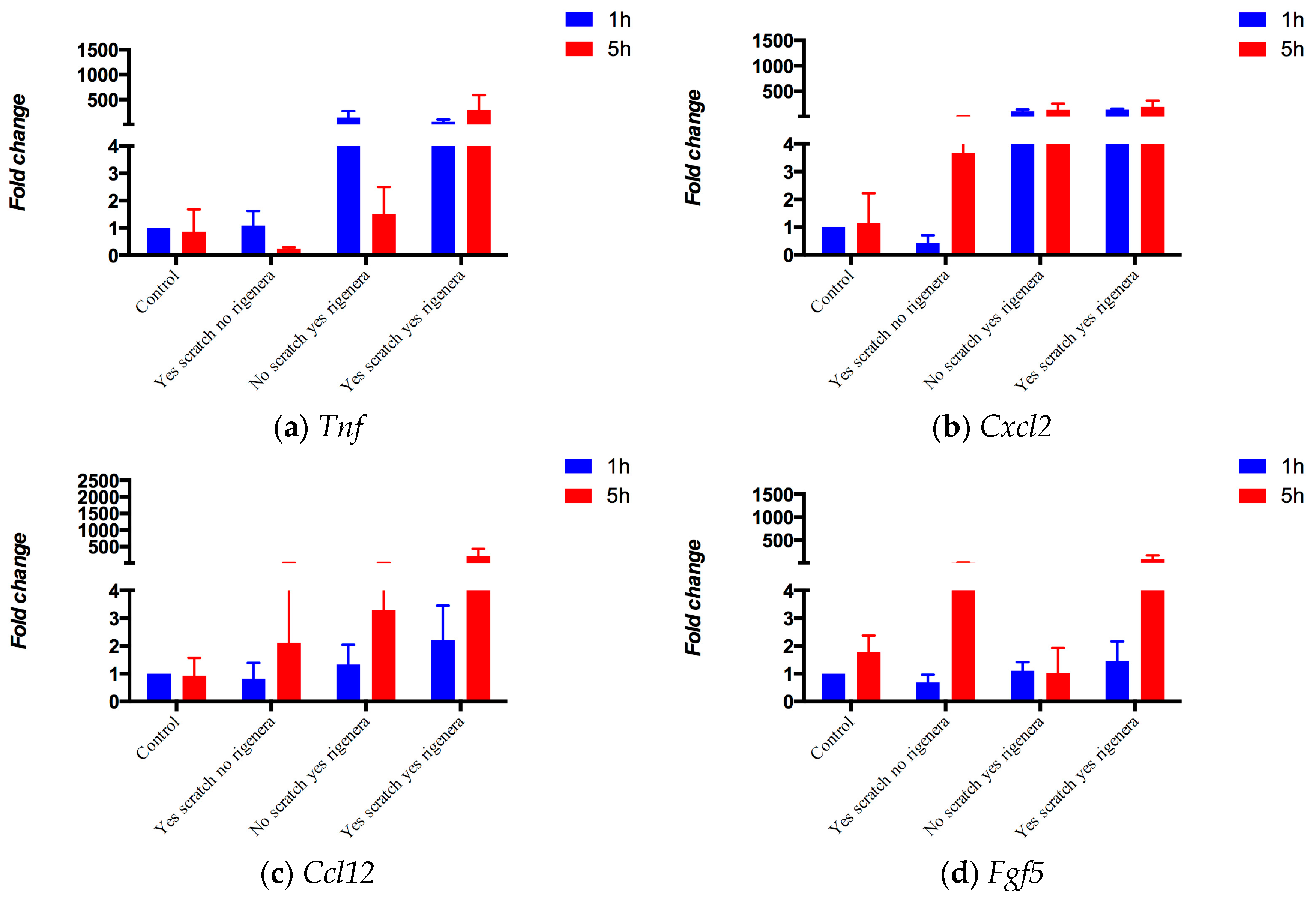
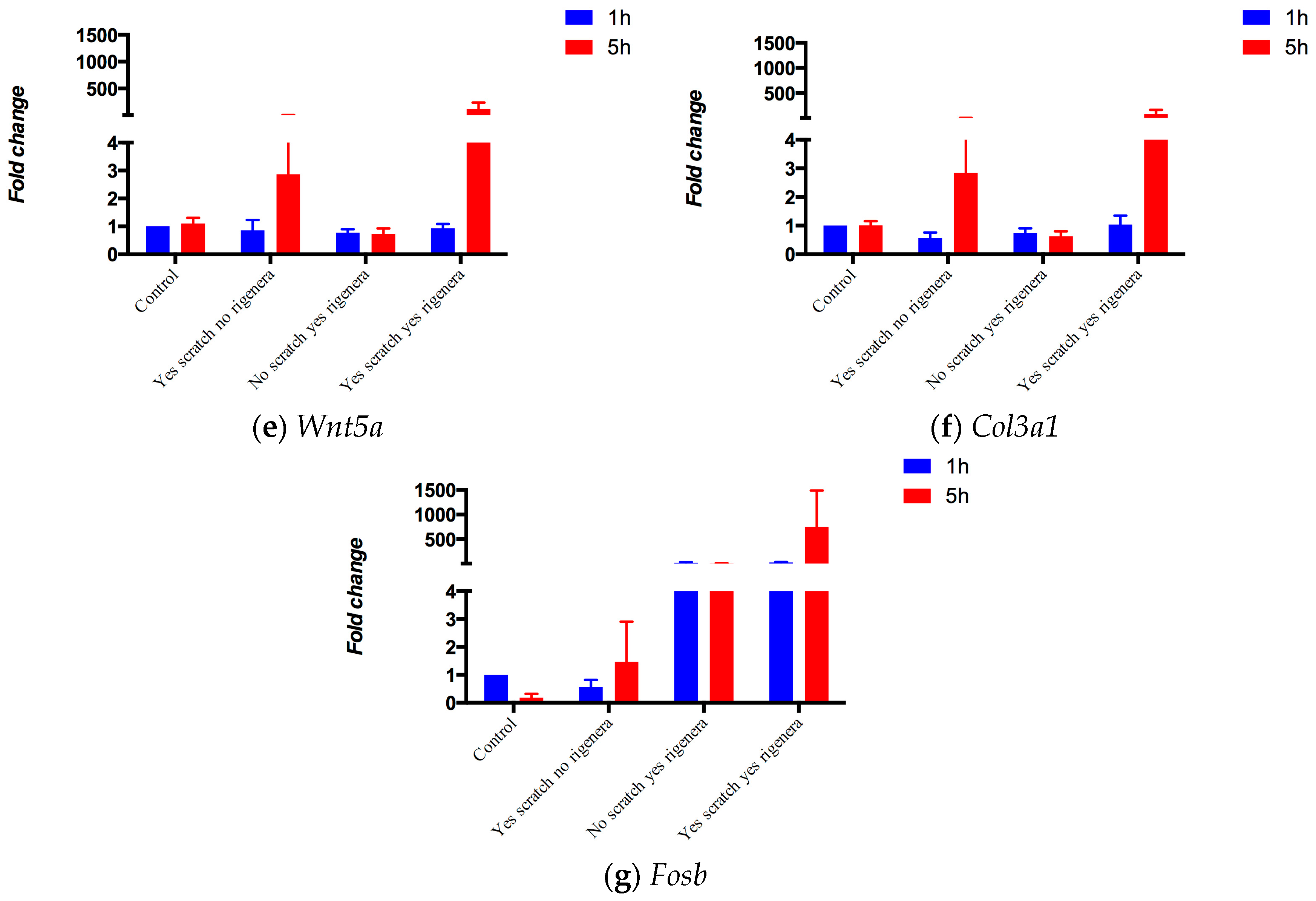
2.1. Assessing the Starting Node Set
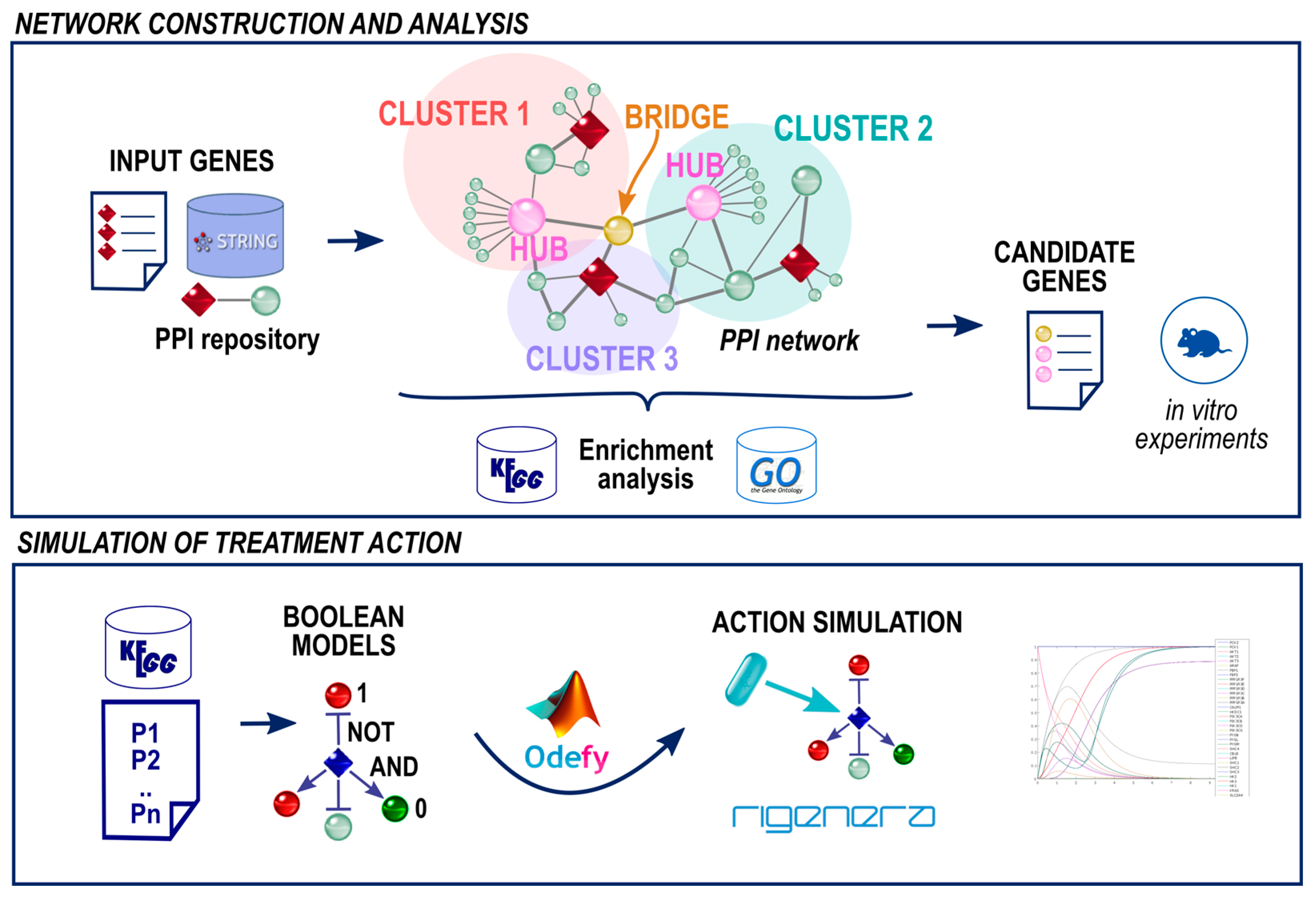
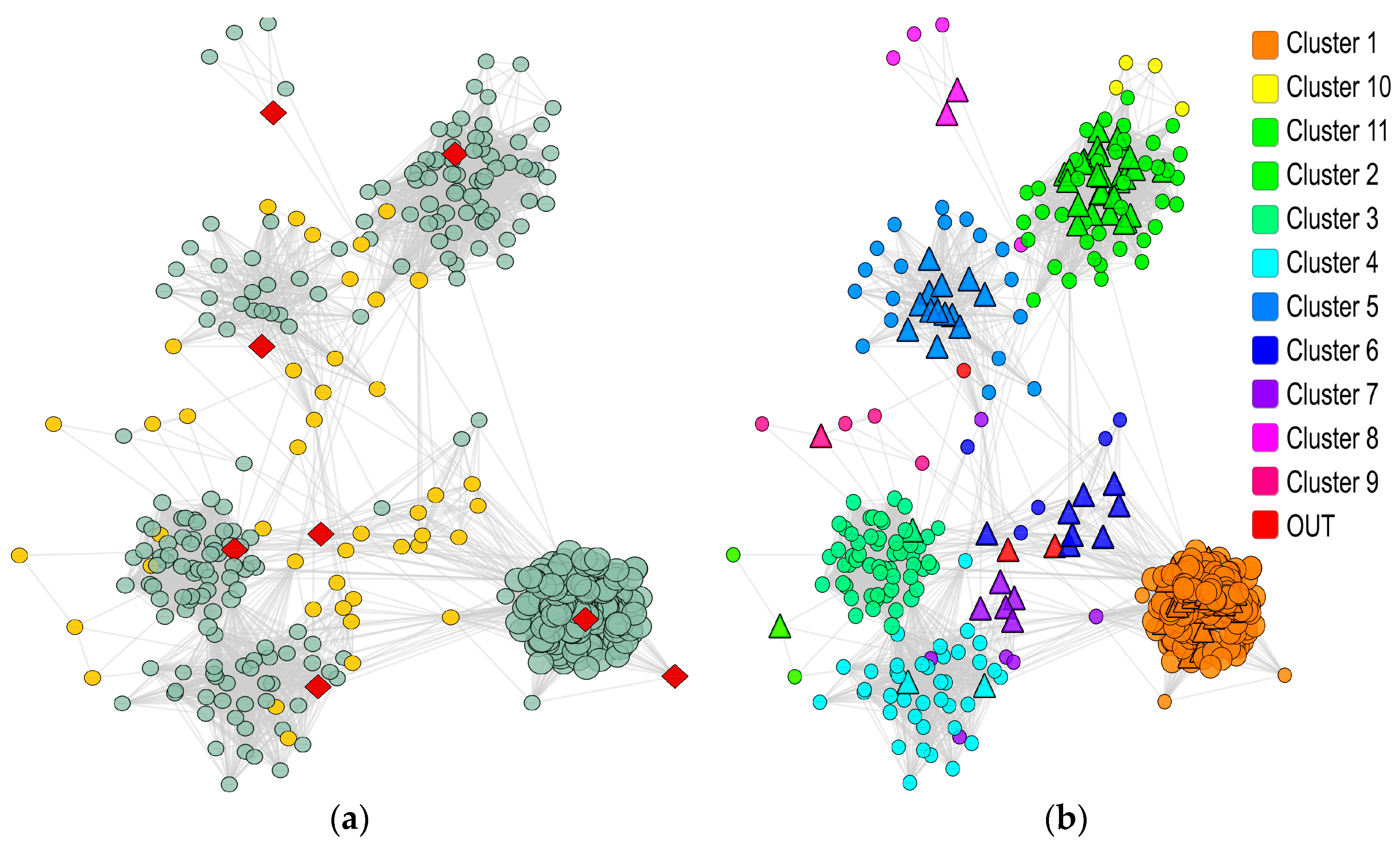
2.2. A Protein Network for Wound Healing
2.3. Pivotal Genes in Wound Healing: Hub Nodes of Clusters as the Key Elements in Regulation
2.4. Pivotal Genes in Wound Healing: Bridge Nodes
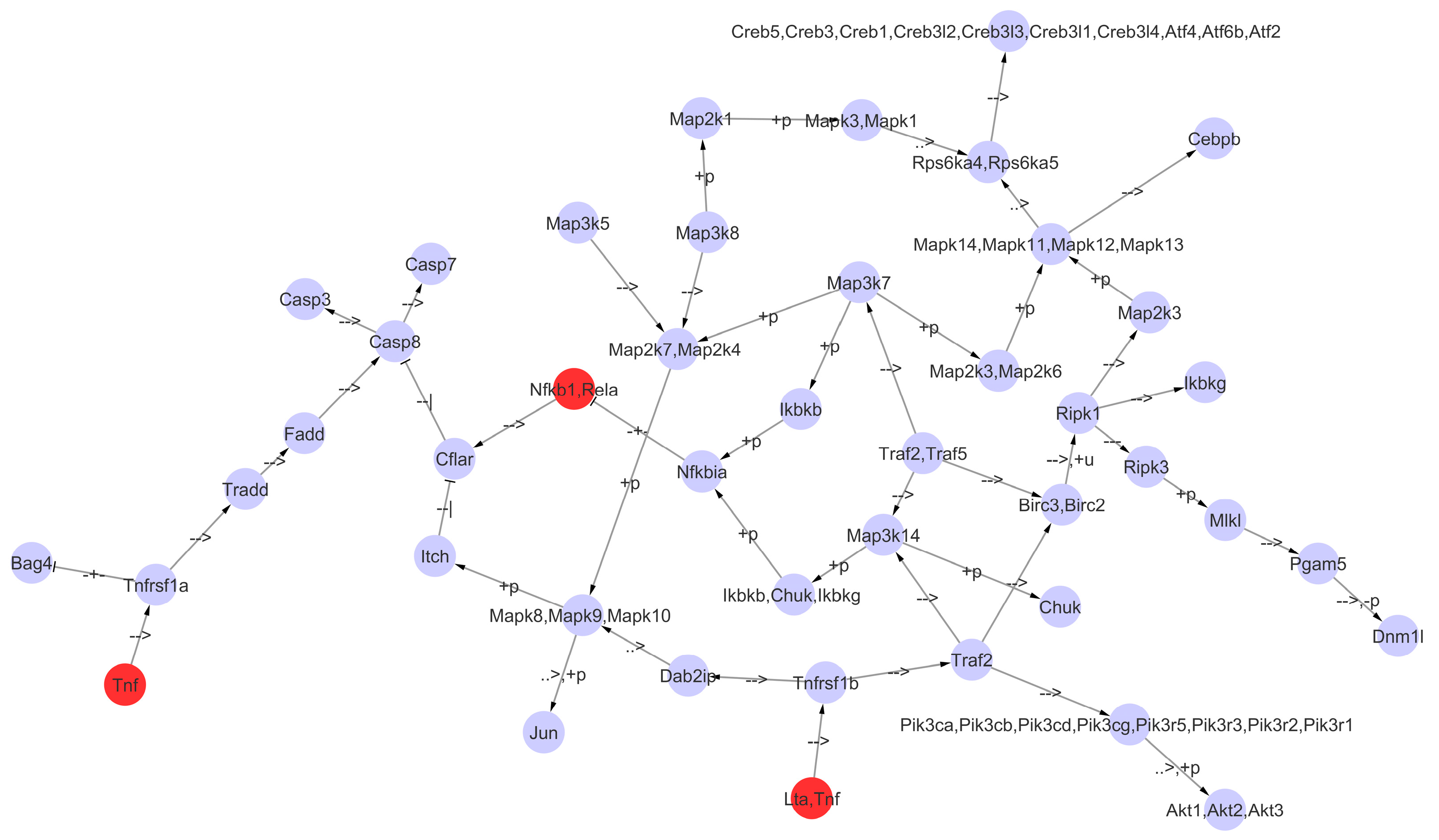
2.5. Simulation of KEGG TNF Signaling Pathway Dynamics
3. Discussion
3.1. Automatically Assembling a Gene Network
3.2. Unveiling Cluster Hub Nodes
- Cluster 1 gathers chemokine/chemotaxis processes, related to cell migration towards chemical gradients. In particular, chemotaxis plays an important role during the inflammatory phase of healing processes [40].
- Cluster 2 is labeled by extracellular matrix/skin development terms.
- Cluster 3 gathers genes involved mainly in DNA regulation of transcription.
- Cluster 4 gathers regenerative processes and cell growth, being labelled by cancer, pluripotency, Wnt, and mTOR signaling pathways.
- Cluster 5, like cluster 2, is characterized by processes of cell adhesion and cell-cell junction formation. This is confirmed by the presence of the Rap1 and Ras signaling pathways, both involved in cell proliferation, survival, growth, migration, differentiation, or cytoskeletal dynamism.
- Cluster 6 is characterized by inflammation and immune response processes. In fact, it is enriched by terms such as MAPKs, TNF signaling pathway, inflammation regulation, and leukocyte migration. The presence of osteoclast proliferation calcium ion terms is consistent with the involvement of this cluster in new bone formation. Although not directly relevant in wound healing, the fact that bone formation terms were grouped in the same cluster indicates how the clustering technique successfully gathered similar genes in a consistent, meaningful fashion.
3.3. Unveiling Bridge Nodes
3.4. A Boolean Network to Study the TNF Signaling Pathway
3.5. Summary
4. Materials and Methods
4.1. Selection of the Input Gene Set
4.1.1. Gene Expression in vitro Experiments
4.2. Protein-Protein Interaction Network Construction
4.2.1. Enrichment analyses
4.3. Identification of Candidate Genes
4.3.1. Cluster Hubs
4.3.2. Bridge Nodes
4.4. How Do Genes React to Rigenera® Stimulus? A Simulation of the Kegg Tnf Signaling Pathway
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sotiriou, C.; Piccart, M.J. Taking gene-expression profiling to the clinic: When will molecular signatures become relevant to patient care? Nat. Rev. Cancer 2007, 7, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Sorg, H.; Tilkorn, D.J.; Hager, S.; Hauser, J.; Mirastschijski, U. Skin wound healing: An update on the current knowledge and concepts. Eur. Surg. Res. 2017, 58, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Maxson, S.; Lopez, E.A.; Yoo, D.; Danilkovitch-Miagkova, A.; Leroux, M.A. Concise review: Role of mesenchymal stem cells in wound repair. Stem Cells Transl. Med. 2012, 1, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Boucek, R.J. Factors affecting wound healing. Otolaryngol. Clin. N. Am. 1984, 17, 243–264. [Google Scholar]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.S.; Mooney, D.J. Regenerative medicine: Current therapies and future directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed]
- Marcarelli, M.; Trovato, L.; Novarese, E.; Riccio, M.; Graziano, A. Rigenera protocol in the treatment of surgical wound dehiscence. Int. Wound J. 2017, 14, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Trovato, L.; Monti, M.; Del Fante, C.; Cervio, M.; Lampinen, M.; Ambrosio, L.; Redi, C.A.; Perotti, C.; Kankuri, E.; Ambrosio, G.; et al. A new medical device rigeneracons allows to obtain viable micro-grafts from mechanical disaggregation of human tissues. J. Cell. Physiol. 2015, 230, 2299–2303. [Google Scholar] [CrossRef] [PubMed]
- Svolacchia, F.; De Francesco, F.; Trovato, L.; Graziano, A.; Ferraro, G.A. An innovative regenerative treatment of scars with dermal micrografts. J. Cosmet. Dermatol. 2016, 15, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Purpura, V.; Bondioli, E.; Graziano, A.; Trovato, L.; Melandri, D.; Ghetti, M.; Marchesini, A.; Cusella De Angelis, M.G.; Benedetti, L.; Ceccarelli, G.; et al. Tissue characterization after a new disaggregation method for skin micro-grafts generation. J. Vis. Exp. 2016, 109, e53579. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, F.; Graziano, A.; Trovato, L.; Ceccarelli, G.; Romano, M.; Marcarelli, M.; Cusella De Angelis, G.M.; Cillo, U.; Riccio, M.; Ferraro, G.A. A regenerative approach with dermal micrografts in the treatment of chronic ulcers. Stem Cell Rev. 2017, 13, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmaros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed]
- Rodius, S.; Androsova, G.; Gotz, L.; Liechti, R.; Crespo, I.; Merz, S.; Nazarov, P.V.; de Klein, N.; Jeanty, C.; Gonzalez-Rosa, J.M.; et al. Analysis of the dynamic co-expression network of heart regeneration in the zebrafish. Sci. Rep. 2016, 6, 26822. [Google Scholar] [CrossRef] [PubMed]
- Elson, D.A.; Ryan, H.E.; Snow, J.W.; Johnson, R.; Arbeit, J.M. Coordinate up-regulation of hypoxia inducible factor (HIF)-1α and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res. 2000, 60, 6189–6195. [Google Scholar] [PubMed]
- Ruthenborg, R.J.; Ban, J.J.; Wazir, A.; Takeda, N.; Kim, J.W. Regulation of wound healing and fibrosis by hypoxia and hypoxia-inducible factor-1. Mol. Cells 2014, 37, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Feezor, R.J.; Paddock, H.N.; Baker, H.V.; Varela, J.C.; Barreda, J.; Moldawer, L.L.; Schultz, G.S.; Mozingo, D.W. Temporal patterns of gene expression in murine cutaneous burn wound healing. Physiol. Genom. 2004, 16, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Bryan, D.; Walker, K.B.; Ferguson, M.; Thorpe, R. Cytokine gene expression in a murine wound healing model. Cytokine 2005, 31, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Qiagen Website. Mouse Wound Healing PCR Array. Available online: http://www.sabiosciences.com/rt_pcr_product/HTML/PAMM-121A.html (accessed on 9 June 2017).
- Ding, J.; Tredget, E.E. The role of chemokines in fibrotic wound healing. Adv. Wound Care (New Rochelle) 2015, 4, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, S.; Matsuo, A.; Yagi, Y.; Ikematsu, K.; Tsuda, R.; Nakasono, I. The time-course analysis of gene expression during wound healing in mouse skin. Leg Med. (Tokyo) 2009, 11, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Chi, L.; Tian, H.; Cai, W.; Sun, C.; Wang, T.; Zhou, X.; Shao, M.; Zhu, Y.; Niu, C.; et al. The activation of the nf-kappab-jnk pathway is independent of the pi3k-rac1-jnk pathway involved in the bfgf-regulated human fibroblast cell migration. J. Dermatol. Sci. 2016, 82, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Houschyar, K.S.; Momeni, A.; Pyles, M.N.; Maan, Z.N.; Whittam, A.J.; Siemers, F. Wnt signaling induces epithelial differentiation during cutaneous wound healing. Organogenesis 2015, 11, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Labus, M.B.; Stirk, C.M.; Thompson, W.D.; Melvin, W.T. Expression of wnt genes in early wound healing. Wound Repair Regen. 1998, 6, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Artlett, C.M.; Blankenhorn, E.P. Tight skin 2 mice exhibit a novel time line of events leading to increased extracellular matrix deposition and dermal fibrosis. Matrix Biol. 2014, 38, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.W.; Wang, Y.; Mauldin, E.A.; Liechty, K.W.; Adams, S.L. Diminished type III collagen promotes myofibroblast differentiation and increases scar deposition in cutaneous wound healing. Cells Tissues Organs 2011, 194, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Crane, N.J.; Brown, T.S.; Evans, K.N.; Hawksworth, J.S.; Hussey, S.; Tadaki, D.K.; Elster, E.A. Monitoring the healing of combat wounds using raman spectroscopic mapping. Wound Repair Regen. 2010, 18, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Indrajana, T.; Spieksma, F.T.; Voorhorst, R. Comparative study of the intracutaneous, scratch and prick tests in allergy. Ann. Allergy 1971, 29, 639–650. [Google Scholar] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The string database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Vitali, F.; Cohen, L.D.; Demartini, A.; Amato, A.; Eterno, V.; Zambelli, A.; Bellazzi, R. A network-based data integration approach to support drug repurposing and multi-target therapies in triple negative breast cancer. PLoS ONE 2016, 11, e0162407. [Google Scholar] [CrossRef] [PubMed]
- Vitali, F.; Mulas, F.; Marini, P.; Bellazzi, R. Network-based target ranking for polypharmacological therapies. J. Biomed. Inform. 2013, 46, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Motenko, H.; Neuhauser, S.B.; O’Keefe, M.; Richardson, J.E. Mousemine: A new data warehouse for mgi. Mamm. Genome 2015, 26, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Gharaee-Kermani, M.; Phan, S.H. Role of cytokines and cytokine therapy in wound healing and fibrotic diseases. Curr. Pharm. Des. 2001, 7, 1083–1103. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shu, B.; Yang, R.; Xu, Y.; Xing, B.; Liu, J.; Chen, L.; Qi, S.; Liu, X.; Wang, P.; et al. Wnt and notch signaling pathway involved in wound healing by targeting c-myc and hes1 separately. Stem Cell Res. Ther. 2015, 6, 120. [Google Scholar] [CrossRef] [PubMed]
- Fathke, C.; Wilson, L.; Shah, K.; Kim, B.; Hocking, A.; Moon, R.; Isik, F. Wnt signaling induces epithelial differentiation during cutaneous wound healing. BMC Cell Biol. 2006, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Vorotnikov, A.V. Chemotaxis: Movement, direction, control. Biochemistry (Moscow) 2011, 76, 1528–1555. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Bergmann, A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [PubMed]
- La Fortezza, M.; Schenk, M.; Cosolo, A.; Kolybaba, A.; Grass, I.; Classen, A.K. JAK/STAT signalling mediates cell survival in response to tissue stress. Development 2016, 143, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Escuin-Ordinas, H.; Li, S.; Xie, M.W.; Sun, L.; Hugo, W.; Huang, R.R.; Jiao, J.; De-Faria, F.M.; Realegeno, S.; Krystofinski, P.; et al. Cutaneous wound healing through paradoxical mapk activation by braf inhibitors. Nat. Commun. 2016, 7, 12348. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, Y.J.; Bian, A.H.; Zuo, H.B.; Zhu, T.W.; Ji, S.X.; Kong, F.; Yin, D.Q.; Wang, C.B.; Wang, Z.F.; et al. The regulatory role of activating transcription factor 2 in inflammation. Mediat. Inflamm. 2014, 2014, 950472. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; McCloskey, D.; Zielinski, D.C.; Sonnenschein, N.; Jamshidi, N.; Palsson, B.O. Personalized whole-cell kinetic models of metabolism for discovery in genomics and pharmacodynamics. Cell Syst. 2015, 1, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.Js: A graph theory library for visualisation and analysis. Bioinformatics 2016, 32, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z. Pubmed and beyond: A survey of web tools for searching biomedical literature. Database (Oxford) 2011, 2011, baq036. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. Genbank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [PubMed]
- Pundir, S.; Martin, M.J.; O’Donovan, C. Uniprot protein knowledgebase. Methods Mol. Biol. 2017, 1558, 41–55. [Google Scholar] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Ruxton, G.D. The unequal variance t-test is an underused alternative to student’s t-test and the mann–Whitney u test. Behav. Ecol. 2006, 17, 688–690. [Google Scholar] [CrossRef]
- Whitlock, M.C. Combining probability from independent tests: The weighted z-method is superior to fisher’s approach. J. Evol. Biol. 2005, 18, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Regan, K.; Huang, Y.; Zhang, Q.; Li, J.; Seiwert, T.Y.; Cohen, E.E.; Xing, H.R.; Lussier, Y.A. Single sample expression-anchored mechanisms predict survival in head and neck cancer. PLoS Comput. Biol. 2012, 8, e1002350. [Google Scholar] [CrossRef] [PubMed]
- Stouffer, S.A. Adjustment during Army Life; Princeton University Press: Princeston, NJ, USA, 1949. [Google Scholar]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. String: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Safari-Alighiarloo, N.; Taghizadeh, M.; Rezaei-Tavirani, M.; Goliaei, B.; Peyvandi, A.A. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol. Hepatol. Bed Bench 2014, 7, 17–31. [Google Scholar] [PubMed]
- Chang, X.; Xu, T.; Li, Y.; Wang, K. Dynamic modular architecture of protein-protein interaction networks beyond the dichotomy of ‘date’ and ‘party’ hubs. Sci. Rep. 2013, 3, 1691. [Google Scholar] [CrossRef] [PubMed]
- Asur, S.; Ucar, D.; Parthasarathy, S. An ensemble framework for clustering protein-protein interaction networks. Bioinformatics 2007, 23, i29–i40. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.; Kotz, D. Localized bridging centrality for distributed network analysis. In Proceedings of the 17th IEEE International Conference on Computer Communications and Networks (ICCCN’08), St. Thomas, U.S. Virgin Islands, USA, 3–7 August 2008; IEEE: Piscataway, NJ, USA, 2008; pp. 1–6. [Google Scholar]
- Ramanathan, M.; Zhang, A.; Cho, Y.-R.; Hwang, W. Bridging Centrality: Identifying Bridging Nodes in Scale-Free Networks. Proceeding of the 12th ACM SIGKDD international conference on Knowlege discovery and data mining (KDD‘06), Philadelphia, PA, USA, 20–23 August 2006. [Google Scholar]
- Valente, T.W.; Fujimoto, K. Bridging: Locating critical connectors in a network. Soc. Netw. 2010, 32, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E. A measure of betweenness centrality based on random walks. Soc. Netw. 2005, 27, 39–54. [Google Scholar] [CrossRef]
- Krumsiek, J.; Polsterl, S.; Wittmann, D.M.; Theis, F.J. Odefy—From discrete to continuous models. BMC Bioinform. 2010, 11, 233. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Uniprot ID | Protein Names | Ref. |
|---|---|---|---|
| Tnf | P06804 | Tumor necrosis factor (Cachectin) (TNF-alpha) (Tumor necrosis factor ligand superfamily member 2) (TNF-a) | [17,18,19,20] |
| Cxcl2 | P10889 | C-X-C motif chemokine 2 (Macrophage inflammatory protein 2) (MIP2) | [18,19,21] |
| Ccl12 | Q62401 | C-C motif chemokine 12 (MCP-1-related chemokine) (Monocyte chemoattractant protein 5) (Monocyte chemotactic protein 5) (MCP-5) (Small-inducible cytokine A12) | [20,22] |
| Fgf5 | P15656 | Fibroblast growth factor 5 (FGF-5) (Heparin-binding growth factor 5) (HBGF-5) | [19,23] |
| Wnt5a | P22725 | Protein Wnt-5a | [20,24,25] |
| Col3a1 | P08121 | Collagen alpha-1(III) chain | [20,26,27,28] |
| Fosb | P13346 | Protein fosB | [22] |
| Pgk1 | P09411 | Phosphoglycerate kinase 1 | [15,16] |
| Cluster | Size | Density | Internal Weight | External Weight | p-Value | # of Hubs |
|---|---|---|---|---|---|---|
| 1 * | 207 | 0.8654 | 1.85 × 104 | 66.97 | <2.2204 × 10−16 ** | 45 |
| 2 * | 66 | 0.5888 | 1263 | 32.96 | <2.2204 × 10−16 ** | 20 |
| 3 * | 58 | 0.8127 | 1343 | 103.4 | <2.2204 × 10−16 ** | 8 |
| 4 * | 42 | 0.6027 | 518.9 | 116.2 | <2.2204 × 10−16 ** | 5 |
| 5 * | 32 | 0.5541 | 274.8 | 23.84 | <2.2204 × 10−16 ** | 13 |
| 6 * | 13 | 0.7173 | 55.95 | 49.28 | 7.27 × 10−5 | 4 |
| 7 | 11 | 0.675 | 37.12 | 77.5 | 0.103808 | 5 |
| 8 * | 6 | 0.54 | 8.1 | 0.8 | 0.00150023 | 2 |
| 9 | 5 | 0.6669 | 6.669 | 7.334 | 0.0712283 | 1 |
| 10 | 4 | 0.9 | 5.4 | 14.4 | 0.997531 | 0 |
| 11 | 3 | 0.5987 | 1.796 | 4.2 | 0.5 | 1 |
| Cluster | # Significant GO Terms | # Significant KEGG Pathways | Top GO Labels (Net Count) | Top KEGG Pathways |
|---|---|---|---|---|
| 1 | 15 | 35 | G-protein coupled receptor signaling pathway chemotaxis C-C chemokine receptor activity | Neuroactive ligand-receptor interaction Chemokine signaling pathway |
| 2 | 23 | 16 | basement membrane external side of plasma membrane extracellular matrix | ECM-receptor interaction Focal adhesion PI3K-Akt signaling pathway |
| 3 | 16 | 2 | positive regulation of transcription negative regulation of transcription regulation of transcription | Adipocytokine signaling pathway Thyroid hormone signaling pathway |
| 4 | 48 | 10 | positive regulation of transcription canonical Wnt signaling pathway Wnt-protein binding | Wnt signaling pathway Breast cancer mTOR signaling pathway |
| 5 | 46 | 20 | positive regulation of cell proliferation lung development cell surface | Rap1 signaling pathway Ras signaling pathway PI3K-Akt signaling pathway |
| 6 | 9 | 53 | regulation of transcription positive regulation of transcription from RNA polymerase II promoter cellular response to calcium ion | Osteoclast differentiation MAPK signaling pathway TNF signaling pathway |
| 8 | 2 | 6 | phosphoglycerate mutase activity glycolytic process | Glycine Glycolysis/Gluconeogenesis Metabolic pathways |
| Gene | GO Term ID | GO Term Name |
|---|---|---|
| Bag4 | GO:0006915 | apoptotic process |
| GO:0010763 | positive regulation of fibroblast migration | |
| GO:0030838 | positive regulation of actin filament polymerization | |
| GO:0042981 | regulation of apoptotic process | |
| GO:0045785 | positive regulation of cell adhesion | |
| GO:0051496 | positive regulation of stress fiber assembly | |
| GO:0071364 | cellular response to epidermal growth factor stimulus | |
| Pik3r1 | GO:0001953 | negative regulation of cell-matrix adhesion |
| GO:0007162 | negative regulation of cell adhesion | |
| GO:0008625 | extrinsic apoptotic signaling pathway via death domain receptors | |
| GO:0043066 | negative regulation of apoptotic process | |
| GO:0030335 | positive regulation of cell migration | |
| Pik3cb | GO:0001935 | endothelial cell proliferation |
| GO:0001952 | regulation of cell-matrix adhesion | |
| GO:0007155 | cell adhesion | |
| GO:0009611 | response to wounding | |
| GO:0030168 | platelet activation | |
| GO:0060055 | angiogenesis involved in wound healing | |
| Map2k6 | GO:0043065 | positive regulation of apoptotic process |
| Map3k7 | GO:0006915 | apoptotic process |
| GO:0016239 | positive regulation of macroautophagy | |
| GO:1902443 | negative regulation of ripoptosome assembly involved in necroptotic process | |
| Mapk10 | GO:0006468 | protein phosphorylation |
| Mapk11 | GO:0006468 | protein phosphorylation |
| GO:0006950 | response to stress | |
| GO:0016310 | phosphorylation | |
| Pik3ca | GO:2000270 | negative regulation of fibroblast apoptotic process |
| GO:0016310 | phosphorylation | |
| Map3k14 | GO:0006468 | protein phosphorylation |
| GO:0006955 | immune response | |
| GO:0016310 | phosphorylation | |
| GO:0030036 | actin cytoskeleton organization | |
| Atf2 | GO:1902110 | positive regulation of mitochondrial membrane permeability involved in apoptotic process |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitali, F.; Marini, S.; Balli, M.; Grosemans, H.; Sampaolesi, M.; Lussier, Y.A.; Cusella De Angelis, M.G.; Bellazzi, R. Exploring Wound-Healing Genomic Machinery with a Network-Based Approach. Pharmaceuticals 2017, 10, 55. https://doi.org/10.3390/ph10020055
Vitali F, Marini S, Balli M, Grosemans H, Sampaolesi M, Lussier YA, Cusella De Angelis MG, Bellazzi R. Exploring Wound-Healing Genomic Machinery with a Network-Based Approach. Pharmaceuticals. 2017; 10(2):55. https://doi.org/10.3390/ph10020055
Chicago/Turabian StyleVitali, Francesca, Simone Marini, Martina Balli, Hanne Grosemans, Maurilio Sampaolesi, Yves A. Lussier, Maria Gabriella Cusella De Angelis, and Riccardo Bellazzi. 2017. "Exploring Wound-Healing Genomic Machinery with a Network-Based Approach" Pharmaceuticals 10, no. 2: 55. https://doi.org/10.3390/ph10020055
APA StyleVitali, F., Marini, S., Balli, M., Grosemans, H., Sampaolesi, M., Lussier, Y. A., Cusella De Angelis, M. G., & Bellazzi, R. (2017). Exploring Wound-Healing Genomic Machinery with a Network-Based Approach. Pharmaceuticals, 10(2), 55. https://doi.org/10.3390/ph10020055