Early Gabapentin Treatment during the Latency Period Increases Convulsive Threshold, Reduces Microglial Activation and Macrophage Infiltration in the Lithium-Pilocarpine Model of Epilepsy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
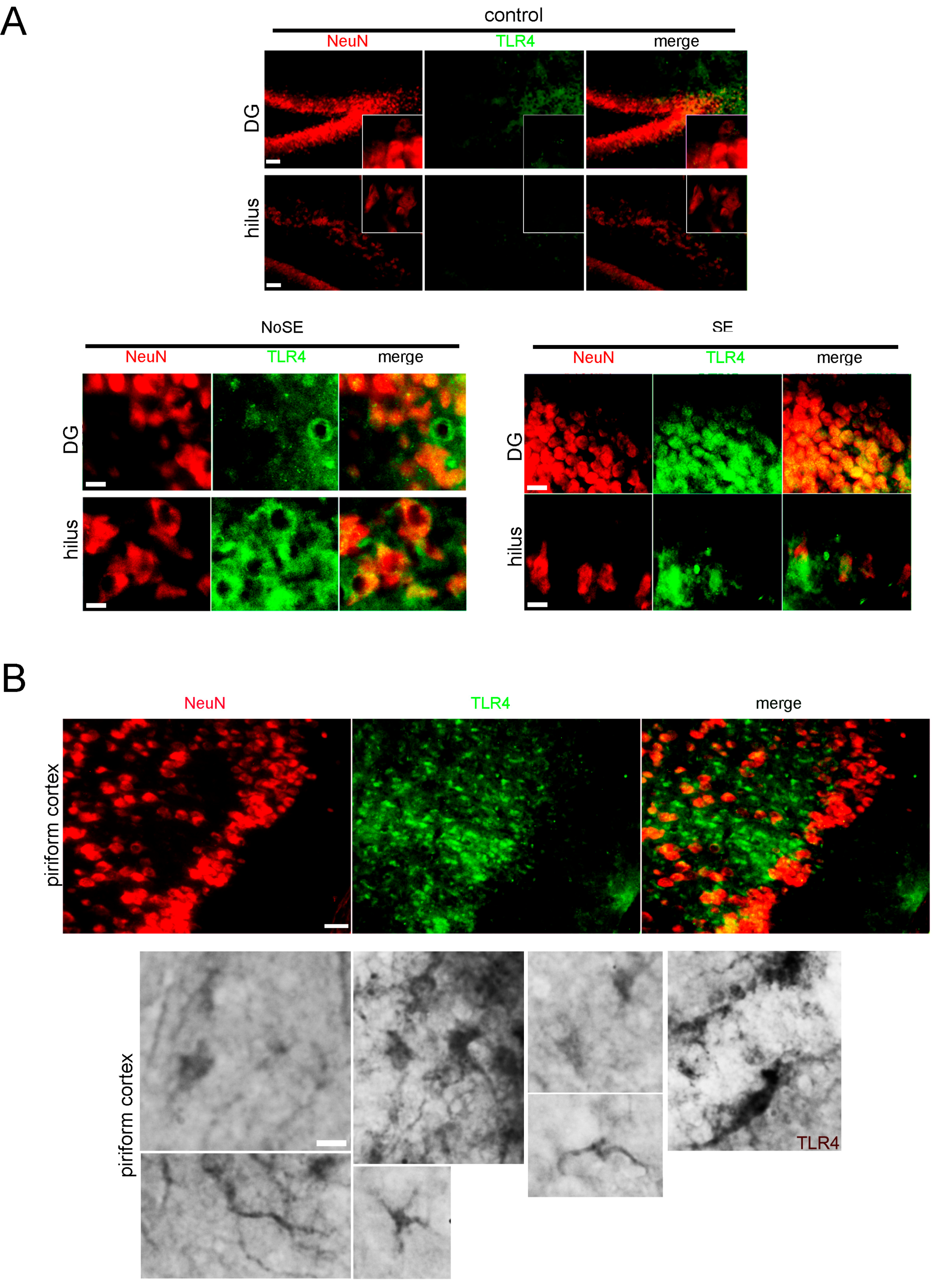
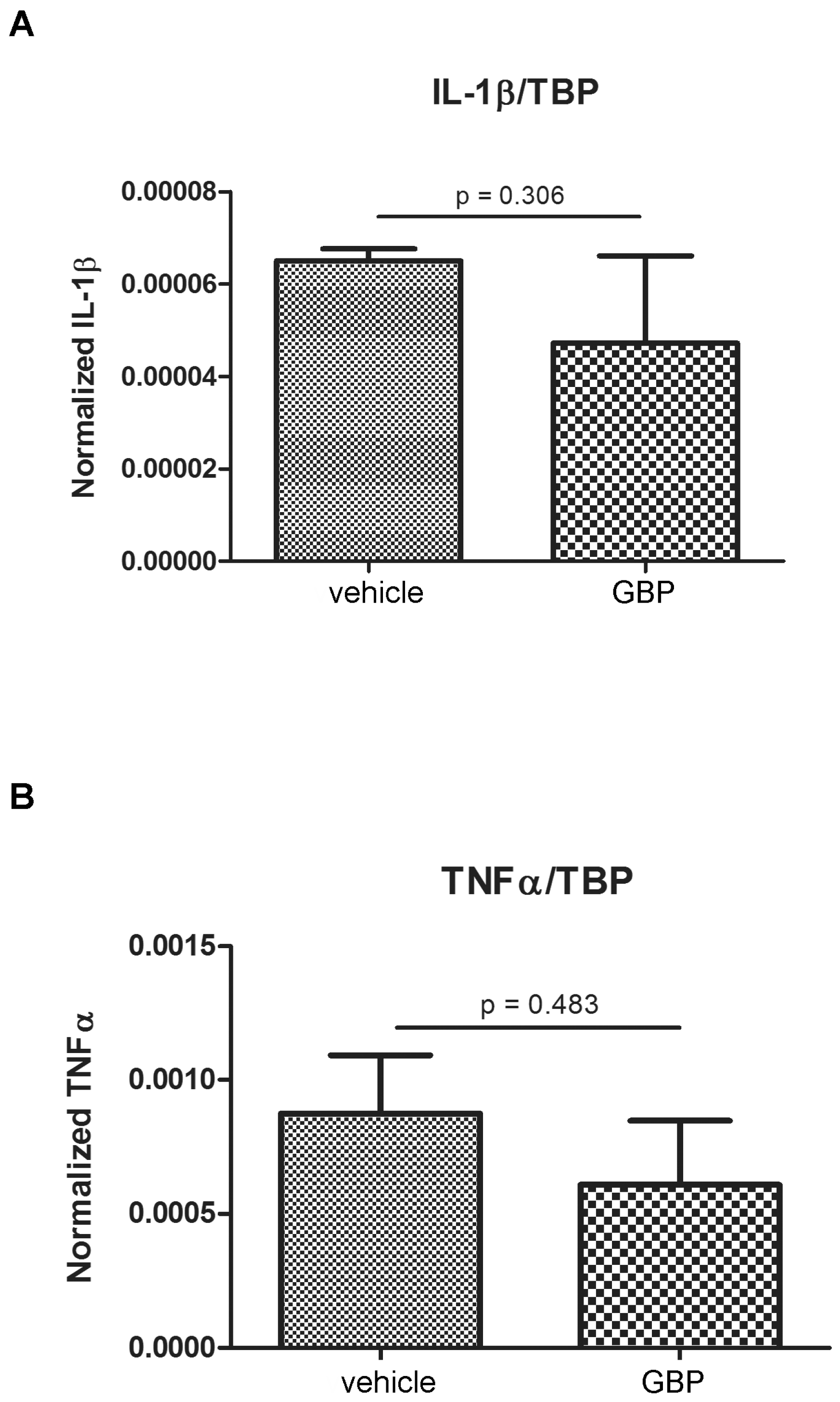
2.1. Higher Expression of Innate Immunity Mediators during the Latency Period after SE
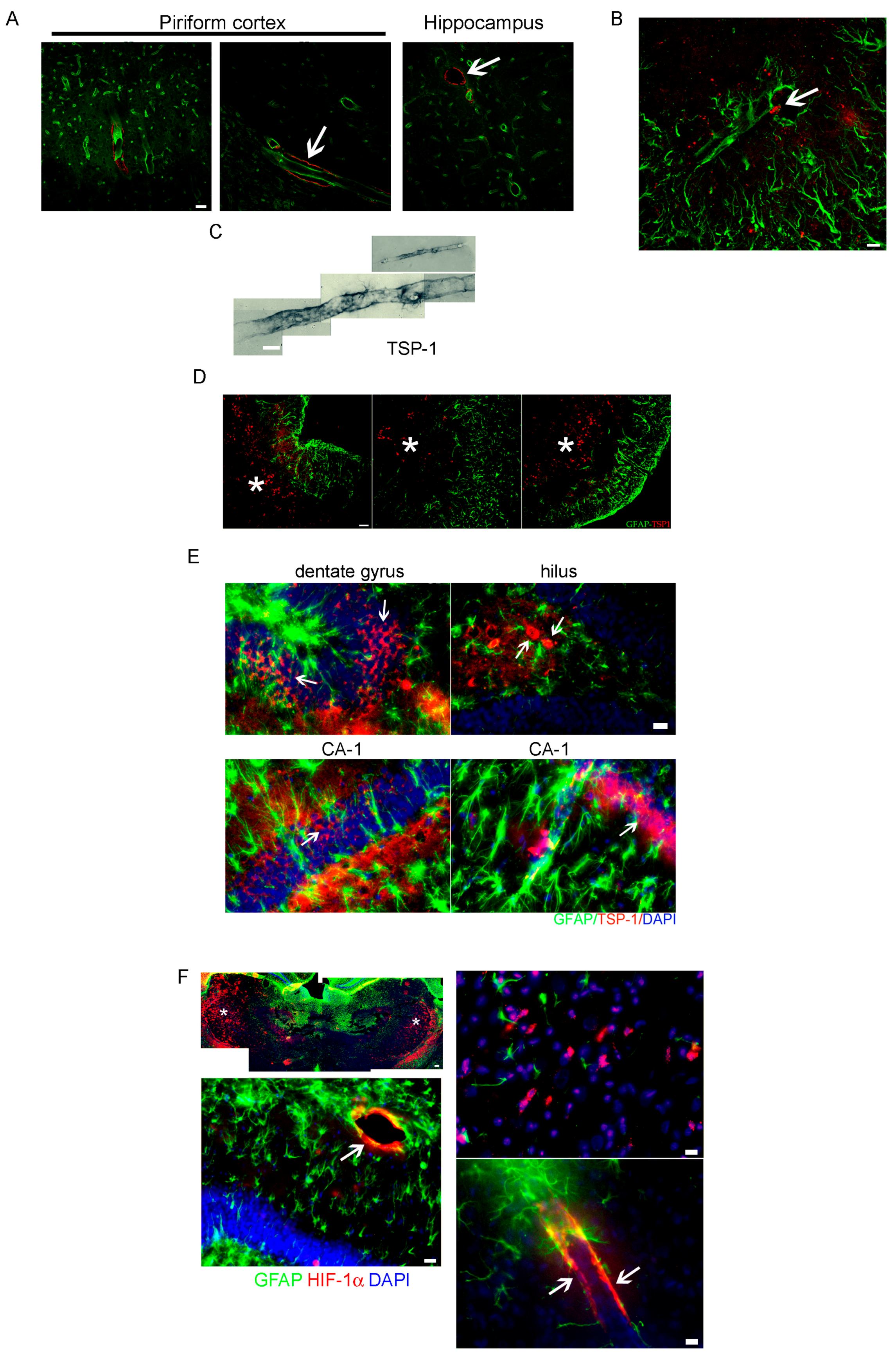
2.2. Altered Blood Brain Barrier (BBB) Permeability during the Latency Period after SE
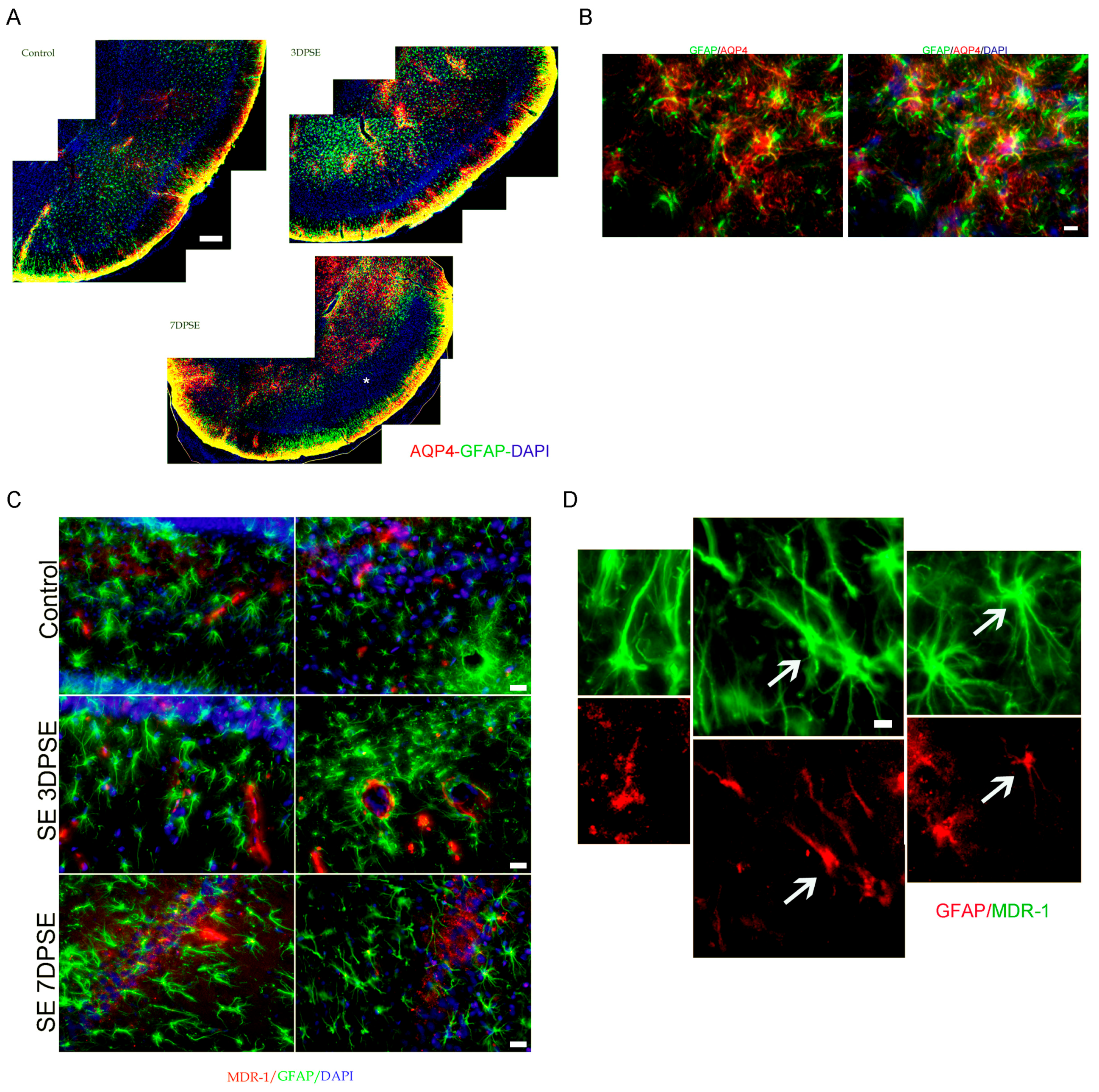
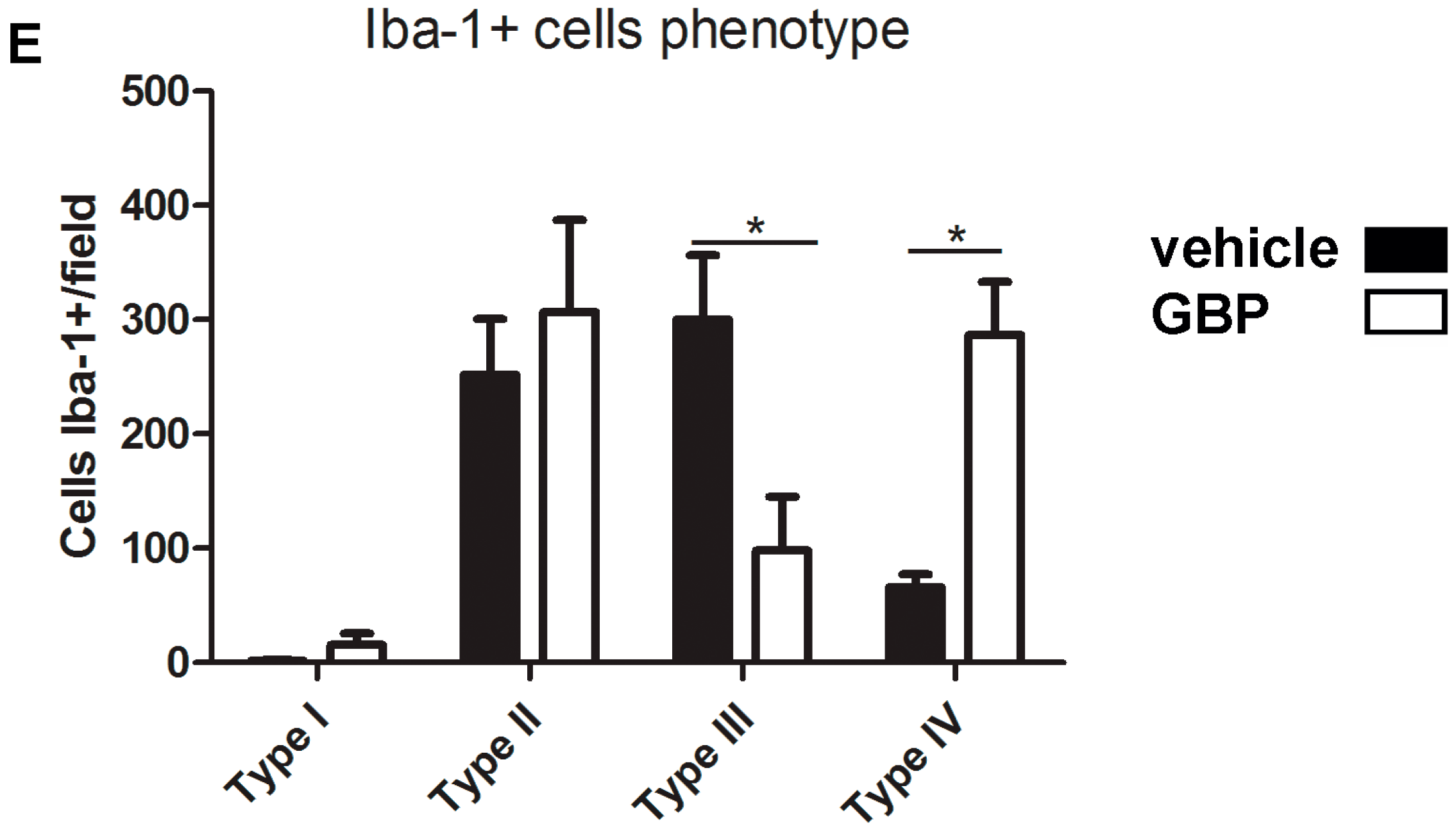
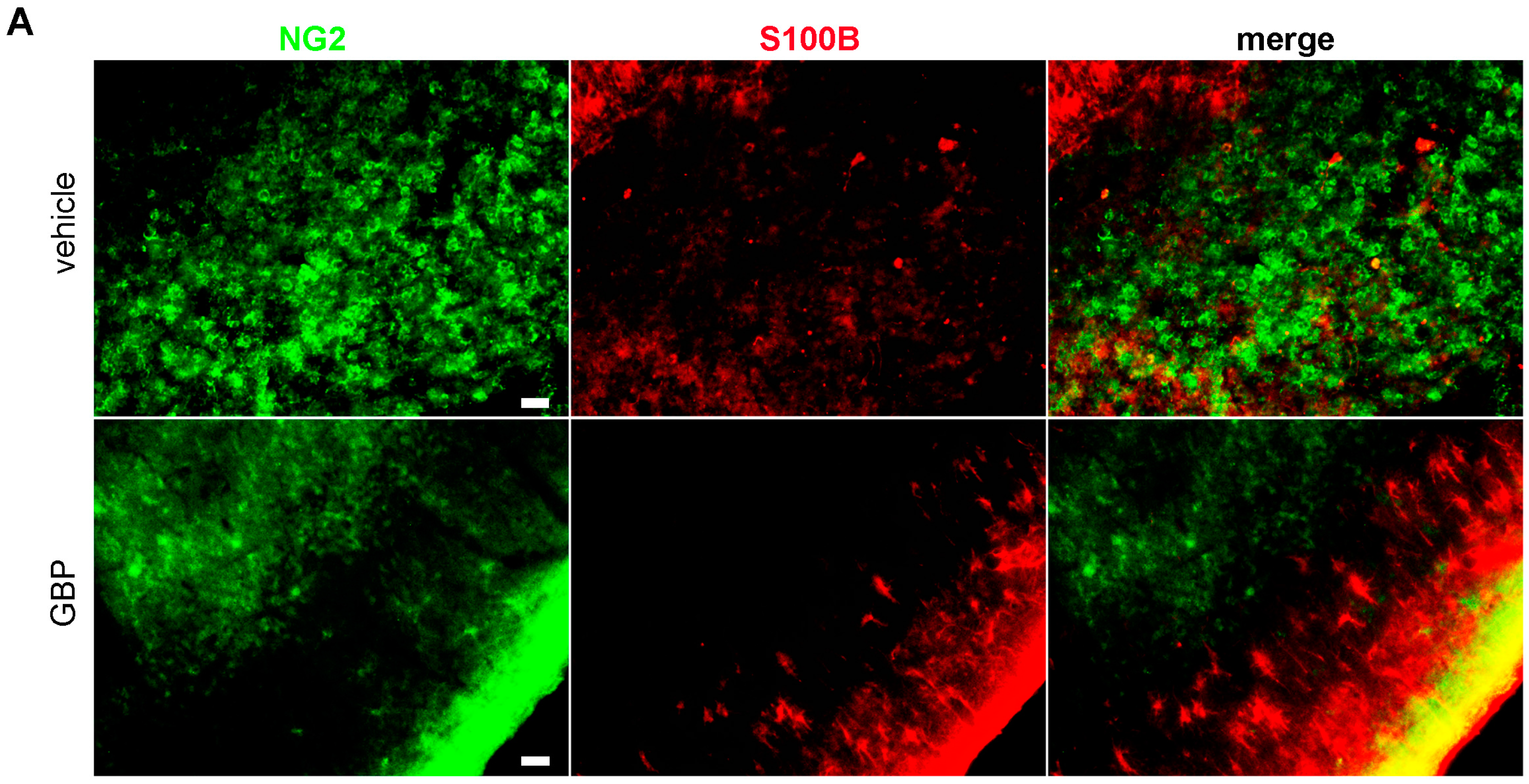
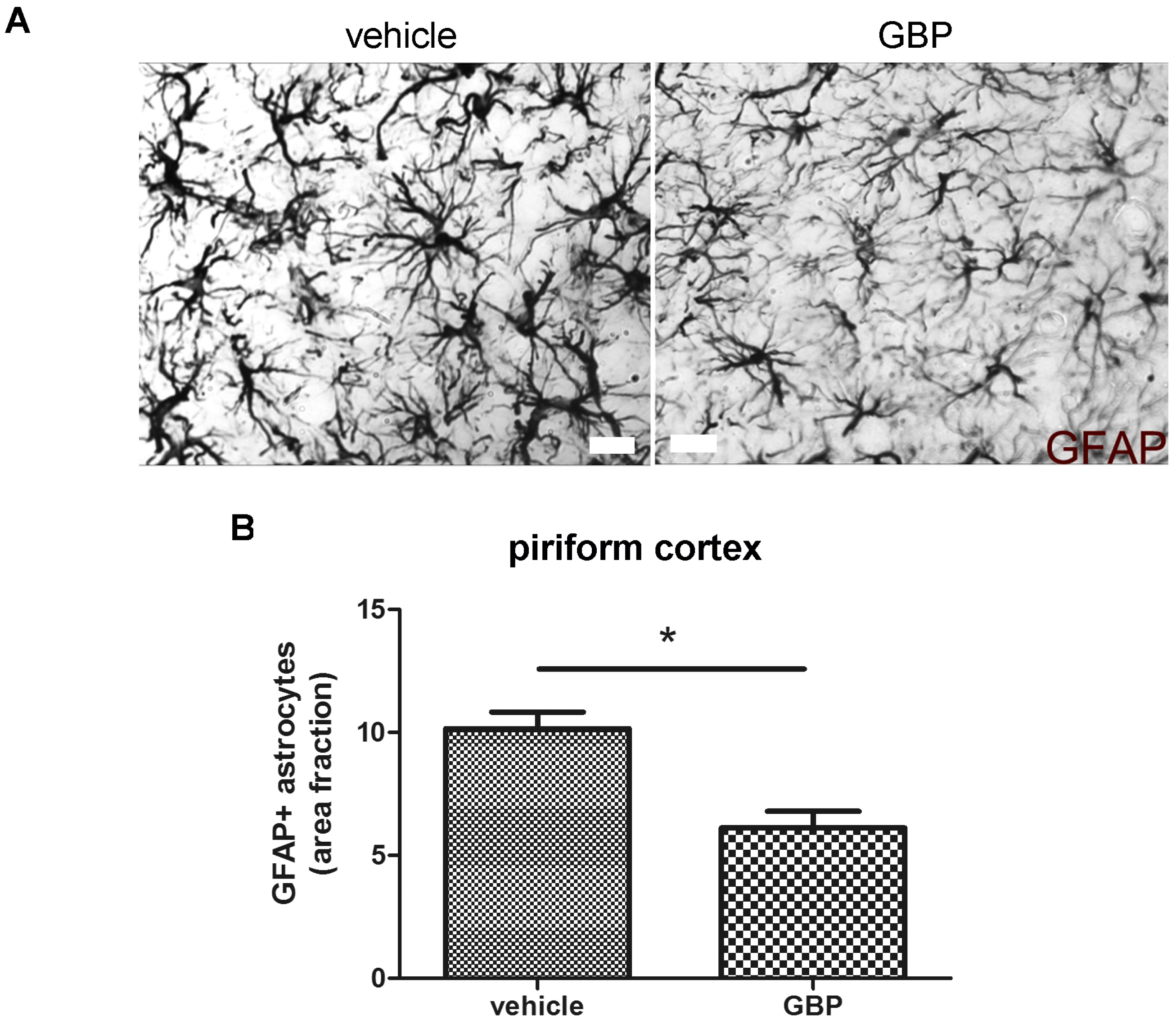
2.3. Early Gabapentin Treatment in the Latency Period Reduces Reactive Gliosis
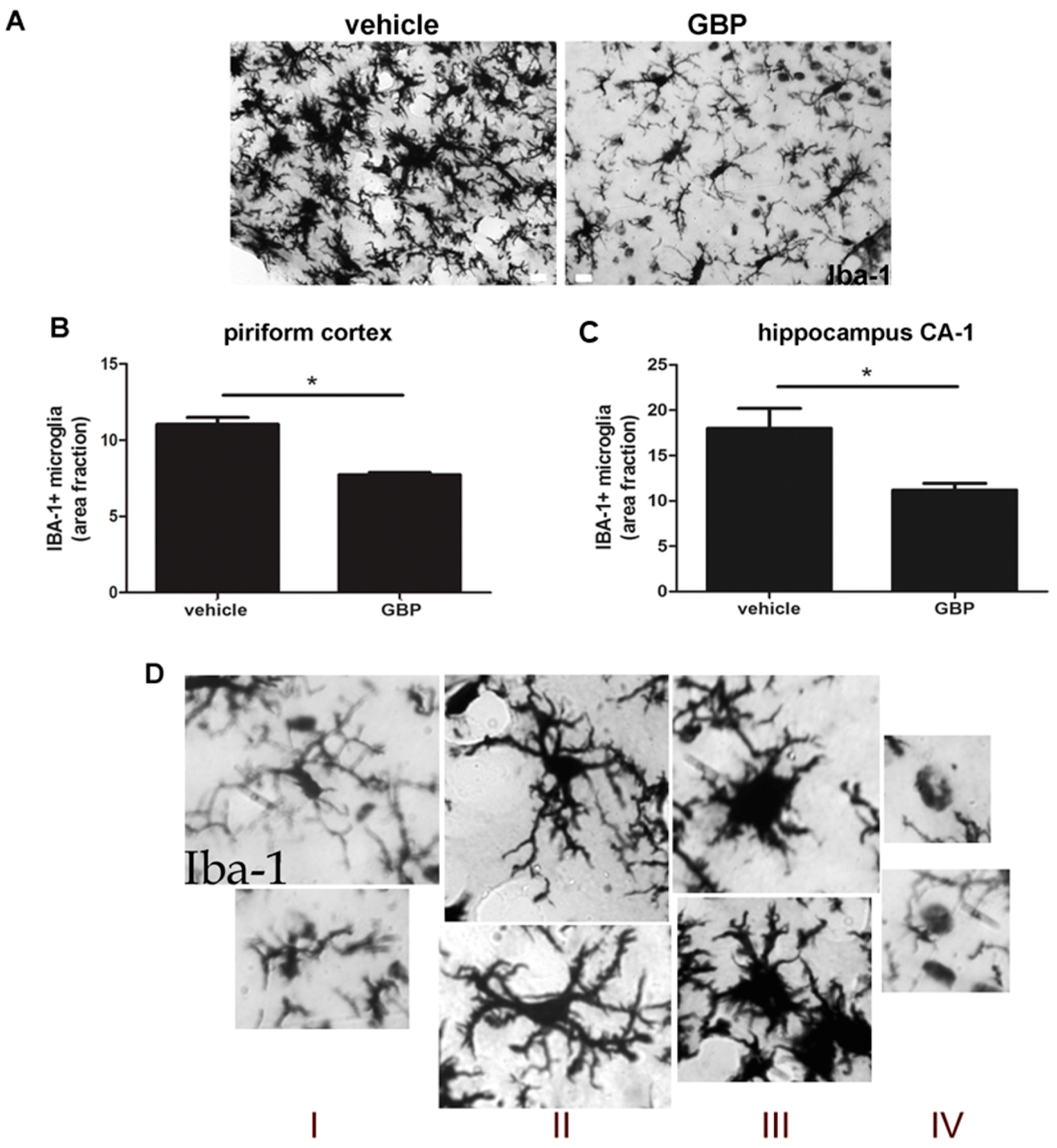
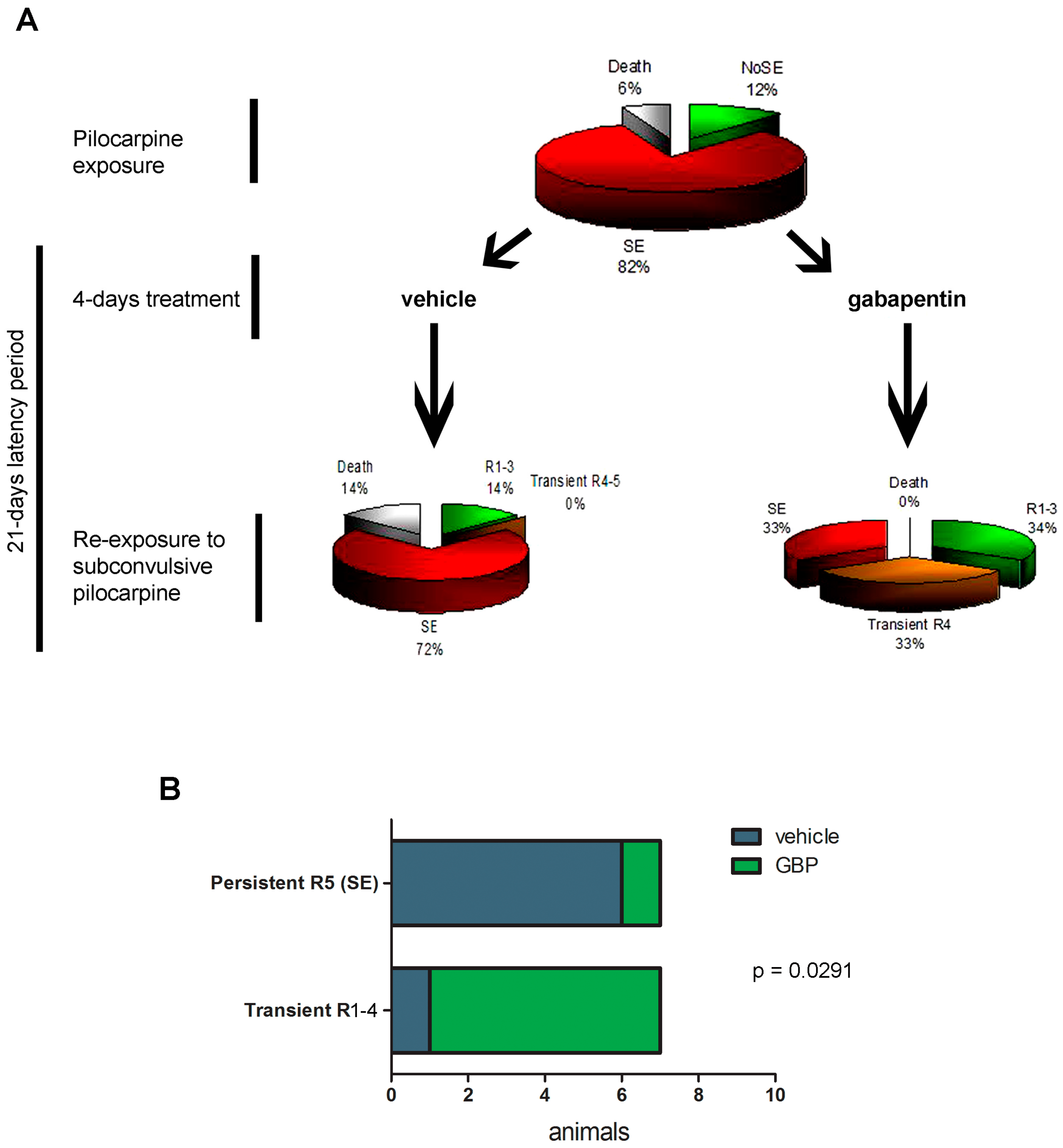
2.4. Gabapentin-Treated Animals Have Increased Convulsive Threshold and Increased Survival When Re-Exposed to Convulsive Agent
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Materials
4.3. Animals and Temporal Lobe Epilepsy Model
4.4. Transfer of eGFP Blood Cells and Bone Marrow-Derived Progenitors
4.5. Immunohistochemistry
4.6. RNA Isolation, Reverse Transcription and Real-Time PCR
4.7. Quantitative Studies
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hesdorffer, D.C.; Beck, V.; Begley, C.E.; Bishop, M.L.; Cushner-Weinstein, S.; Holmes, G.L.; Shafer, P.O.; Sirven, J.I.; Austin, J.K. Research implications of the Institute of Medicine Report, Epilepsy across the Spectrum: Promoting Health and Understanding. Epilepsia 2013, 54, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Clossen, B.L.; Reddy, D.S. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim. Biophys. Acta 2017, 1863, 1519–1538. [Google Scholar] [CrossRef] [PubMed]
- Witt, J.A.; Hoppe, C.; Helmstaedter, C. Neuropsychologist’s (re-)view: Resective versus ablative amygdalohippocampectomies. Epilepsy Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cukiert, A. Vagus Nerve Stimulation for Epilepsy: An Evidence-Based Approach. Prog. Neurol. Surg. 2015, 29, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.; Prasad, A.N. Natural history of temporal lobe epilepsy: Antecedents and progression. Epilepsy Res. Treat. 2012, 2012, 195073. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Lucchi, C.; Vinet, J.; Gualtieri, F.; Marinelli, C.; Torsello, A.; Costantino, L.; Biagini, G. Pathophysiogenesis of mesial temporal lobe epilepsy: Is prevention of damage antiepileptogenic? Curr. Med. Chem. 2014, 21, 663–688. [Google Scholar] [CrossRef] [PubMed]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Maroso, M.; Zardoni, D.; Noé, F.; Ravizza, T. ICE/caspase 1 inhibitors and IL-1beta receptor antagonists as potential herapeutics in epilepsy. Curr. Opin. Investig. Drugs 2010, 11, 43–50. [Google Scholar] [PubMed]
- Kim, S.K.; Nabekura, J.; Koizumi, S. Astrocyte-mediated synapse remodeling in the pathological brain. Glia 2017, 65, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Qin, H.; Chen, J.; Mou, L.; He, Y.; Yan, Y.; Zhou, H.; Lv, Y.; Chen, Z.; Wang, J.; et al. Postnatal activation of TLR4 in astrocytes promotes excitatory synaptogenesis in hippocampal neurons. J. Cell Biol. 2016, 215, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Faria, L.C.; Gu, F.; Parada, I.; Barres, B.; Luo, Z.D.; Prince, D.A. Epileptiform activity and behavioral arrests in mice overexpressing the calcium channel subunit α2δ-1. Neurobiol. Dis. 2017, 102, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Gui, W.; He, H.Y.; Wang, X.S.; Zuo, J.; Huang, L.; Zhou, N.; Wang, K.; Wang, Y. Neuronal and astroglial TGFβ-Smad3 signaling pathways differentially regulate dendrite growth and synaptogenesis. NeuroMol. Med. 2014, 16, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Hawrylak, N.; Chang, F.L.; Greenough, W.T. Astrocytic and synaptic response to kindling in hippocampal subfield CA1. II. Synaptogenesis and astrocytic process increases to in vivo kindling. Brain Res. 1993, 603, 309–316. [Google Scholar] [CrossRef]
- Niquet, J.; Jorquera, I.; Ben-Ari, Y.; Represa, A. NCAM immunoreactivity on mossy fibers and reactive astrocytes in the hippocampus of epileptic rats. Brain Res. 1993, 626, 106–116. [Google Scholar] [CrossRef]
- Lehtimäki, K.A.; Keränen, T.; Palmio, J.; Peltola, J. Levels of IL-1beta and IL-1ra in cerebrospinal fluid of human patients after single and prolonged seizures. Neuroimmunomodulation 2010, 17, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Yang, F.; Wang, Y.G.; Wang, J.C.; Ma, L.; Jiang, W. Brain recruitment of dendritic cells following Li-pilocarpine induced status epilepticus in adult rats. Brain Res. Bull. 2013, 91, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O’Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Celli, R.; Santolini, I.; Guiducci, M.; van Luijtelaar, G.; Parisi, P.; Striano, P.; Gradini, R.; Battaglia, G.; Ngomba, R.T.; Nicoletti, F. The α2δ Subunit and Absence Epilepsy: Beyond Calcium Channels? Curr. Neuropharmacol. 2017, 15, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Okada-Tsuchioka, M.; Segawa, M.; Kajitani, N.; Hisaoka-Nakashima, K.; Shibasaki, C.; Morinobu, S.; Takebayashi, M. Electroconvulsive seizure induces thrombospondin-1 in the adult rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Graber, K.D.; Jin, S.; McDonald, W.; Barres, B.A.; Prince, D.A. Gabapentin decreases epileptiform discharges in a chronic model of neocortical trauma. Neurobiol. Dis. 2012, 48, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Xu, B.; Li, S.S.; Zhang, W.S.; Xu, H.; Deng, X.M.; Zhang, Y.Q. Gabapentin reduces CX3CL1 signaling and blocks spinal microglial activation in monoarthritic rats. Mol. Brain 2012, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.R.; Angelo, M.F.; Villarreal, A.; Lukin, J.; Ramos, A.J. Gabapentin administration reduces reactive gliosis and neurodegeneration after pilocarpine-induced status epilepticus. PLoS ONE 2013, 8, e78516. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualtieri, F.; Marinelli, C.; Longo, D.; Pugnaghi, M.; Nichelli, P.F.; Meletti, S.; Biagini, G. Hypoxia markers are expressed in interneurons exposed to recurrent seizures. Neuromol. Med. 2013, 15, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, C.; Vinet, J.; Meletti, S.; Biagini, G. Ischemic-hypoxic mechanisms leading to hippocampal dysfunction as a consequence of status epilepticus. Epilepsy Behav. 2015, 49, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Serwanski, D.R.; Miralles, C.P.; Fiondella, C.G.; Loturco, J.J.; Rubio, M.E.; De Blas, A.L. Synaptic and nonsynaptic localization of protocadherin-gammaC5 in the rat brain. J. Comp. Neurol. 2010, 518, 3439–3463. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Masià, D.; Díez, I.; Calatayud, S.; Hernández, C.; Cosín-Roger, J.; Hinojosa, J.; Esplugues, J.V.; Barrachina, M.D. Induction of CD36 and thrombospondin-1 in macrophages by hypoxia-inducible factor 1 and its relevance in the inflammatory process. PLoS ONE 2012, 7, e48535. [Google Scholar] [CrossRef] [PubMed]
- Osada-Oka, M.; Ikeda, T.; Akiba, S.; Sato, T. Hypoxia stimulates the autocrine regulation of migration of vascular smooth muscle cells via HIF-1alpha-dependent expression of thrombospondin-1. J. Cell. Biochem. 2008, 104, 1918–1926. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Zhu, G.Y.; Wang, X.; Shi, L.; Du, T.T.; Liu, D.F.; Liu, Y.Y.; Jiang, Y.; Zhang, X.; Zhang, J.G. Anterior thalamic nuclei deep brain stimulation reduces disruption of the blood-brain barrier, albumin extravasation, inflammation and apoptosis in kainic acid-induced epileptic rats. Neurol. Res. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Michalak, Z.; Sano, T.; Engel, T.; Miller-Delaney, S.F.; Lerner-Natoli, M.; Henshall, D.C. Spatio-temporally restricted blood-brain barrier disruption after intra-amygdala kainic acid-induced status epilepticus in mice. Epilepsy Res. 2013, 103, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Teng, Q.; Ghosh, C.; Fan, Q.; Nguyen, M.T.; Desai, N.K.; Bawa, H.; Rasmussen, P.; Masaryk, T.K.; Janigro, D. Blood-brain barrier damage, but not parenchymal White blood cells, is a hallmark of seizure activity. Brain Res. 2010, 1353, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Fabene, P.F.; Navarro Mora, G.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, A.; Ramos, A.J.; García-Rivello, H.; Brusco, A.; Girardi, E. Neuronal and glial expression of the multidrug resistance gene product in an experimental epilepsy model. Cell. Mol. Neurobiol. 2004, 24, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Pekcec, A.; Soldner, E.L.; Zhong, Y.; Schlichtiger, J.; Bauer, B. P-gp Protein Expression and Transport Activity in Rodent Seizure Models and Human Epilepsy. Mol. Pharm. 2017, 14, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Benakis, C.; Garcia-Bonilla, L.; Iadecola, C.; Anrather, J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front. Cell. Neurosci. 2015, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Vizuete, A.F.K.; Hennemann, M.M.; Gonçalves, C.A.; de Oliveira, D.L. Phase-Dependent Astroglial Alterations in Li-Pilocarpine-Induced Status Epilepticus in Young Rats. Neurochem. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Cav3.1 T-type calcium channel modulates the epileptogenicity of hippocampal seizures in the kainic acid-induced temporal lobe epilepsy model. Brain Res. 2015, 1622, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Soukupová, M.; Binaschi, A.; Falcicchia, C.; Zucchini, S.; Roncon, P.; Palma, E.; Magri, E.; Grandi, E.; Simonato, M. Impairment of GABA release in the hippocampus at the time of the first spontaneous seizure in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2014, 257, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, M.; Binaschi, A.; Falcicchia, C.; Palma, E.; Roncon, P.; Zucchini, S.; Simonato, M. Increased extracellular levels of glutamate in the hippocampus of chronically epileptic rats. Neuroscience 2015, 301, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, M.M.; Menegola, M.; Vacher, H.; Rhodes, K.J.; Trimmer, J.S. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 2008, 156, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Suleymanova, E.M.; Shangaraeva, V.A.; van Rijn, C.M.; Vinogradova, L.V. The cannabinoid receptor agonist WIN55.212 reduces consequences of status epilepticus in rats. Neuroscience 2016, 334, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Tchekalarova, J.; Petkova, Z.; Pechlivanova, D.; Moyanova, S.; Kortenska, L.; Mitreva, R.; Lozanov, V.; Atanasova, D.; Lazarov, N.; Stoynev, A. Prophylactic treatment with melatonin after status epilepticus: Effects on epileptogenesis, neuronal damage, and behavioral changes in a kainate model of temporal lobe epilepsy. Epilepsy Behav. 2013, 27, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.D.; Oliveira, R.W.; Futuro Neto, H.d.A.; Nakamura-Palacios, E.M. The role of magnesium sulfate in prevention of seizures induced by pentylenetetrazole in rats. Arq. Neuro-Psiquiatr. 2011, 69, 349–355. [Google Scholar] [CrossRef]
- Biagini, G.; Longo, D.; Baldelli, E.; Zoli, M.; Rogawski, M.A.; Bertazzoni, G.; Avoli, M. Neurosteroids and epileptogenesis in the pilocarpine model: Evidence for a relationship between P450scc induction and length of the latent period. Epilepsia 2009, 50 (Suppl. 1), 53–58. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.; Rustichelli, C.; Curia, G.; Vinet, J.; Lucchi, C.; Pugnaghi, M.; Meletti, S. Neurosteroids and epileptogenesis. J. Neuroendocrinol. 2013, 25, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Majores, M.; Schoch, S.; Lie, A.; Becker, A.J. Molecular neuropathology of temporal lobe epilepsy: Complementary approaches in animal models and human disease tissue. Epilepsia 2007, 48 (Suppl. 2), 4–12. [Google Scholar] [CrossRef] [PubMed]
- Retchkiman, I.; Fischer, B.; Platt, D.; Wagner, A.P. Seizure induced C-Fos mRNA in the rat brain: Comparison between young and aging animals. Neurobiol. Aging 1996, 17, 41–44. [Google Scholar] [CrossRef]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef] [PubMed]
- Vinet, J.; Vainchtein, I.D.; Spano, C.; Giordano, C.; Bordini, D.; Curia, G.; Dominici, M.; Boddeke, H.W.; Eggen, B.J.; Biagini, G. Microglia are less pro-inflammatory than myeloid infiltrates in the hippocampus of mice exposed to status epilepticus. Glia 2016, 64, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Yang, X.L.; Ma, Y.S.; Wei, Y.J.; Yang, M.H.; Chen, X.; Chen, B.; He, Q.; Yang, Q.W.; Yang, H.; et al. TRIF contributes to epileptogenesis in temporal lobe epilepsy during TLR4 activation. Brain Behav. Immun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, J.; Shang, Y.; Zhao, L.; Wang, M.; Shi, J.; Li, S. HMGB1-TLR4 Axis Plays a Regulatory Role in the Pathogenesis of Mesial Temporal Lobe Epilepsy in Immature Rat Model and Children via the p38MAPK Signaling Pathway. Neurochem. Res. 2017, 42, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Pernhorst, K.; Herms, S.; Hoffmann, P.; Cichon, S.; Schulz, H.; Sander, T.; Schoch, S.; Becker, A.J.; Grote, A. TLR4, ATF-3 and IL8 inflammation mediator expression correlates with seizure frequency in human epileptic brain tissue. Seizure 2013, 22, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Iori, V.; Iyer, A.M.; Ravizza, T.; Beltrame, L.; Paracchini, L.; Marchini, S.; Cerovic, M.; Hill, C.; Ferrari, M.; Zucchetti, M.; et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol. Dis. 2017, 99, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Rosciszewski, G.; Cadena, V.; Murta, V.; Lukin, J.; Villarreal, A.; Roger, T.; Ramos, A.J. Toll-Like Receptor 4 (TLR4) and Triggering Receptor Expressed on Myeloid Cells-2 (TREM-2) Activation Balance Astrocyte Polarization into a Proinflammatory Phenotype. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.L.; Puskar, A.; Hoffman, G.E.; Murphy, A.Z.; Saraswati, M.; Fiskum, G. Physiologic progesterone reduces mitochondrial dysfunction and hippocampal cell loss after traumatic brain injury in female rats. Exp. Neurol. 2006, 197, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.T.; Eroglu, C. Molecular mechanisms of astrocyte-induced synaptogenesis. Curr. Opin. Neurobiol. 2017, 45, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009, 158, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Janigro, D. Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia 2012, 53 (Suppl. 1), 26–34. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Szu, J.I.; Binder, D.K. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res. Bull. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Szu, J.I.; Yonan, J.M.; Binder, D.K. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp. Neurol. 2016, 283 Pt A, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Alvestad, S.; Hammer, J.; Hoddevik, E.H.; Skare, Ø.; Sonnewald, U.; Amiry-Moghaddam, M.; Ottersen, O.P. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 2013, 105, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Hsu, M.S.; Seldin, M.M.; Arellano, J.L.; Binder, D.K. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp. Neurol. 2012, 235, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Yeo, S.I.; Ryu, H.J.; Kim, M.J.; Kim, D.S.; Jo, S.M.; Kang, T.C. Astroglial loss and edema formation in the rat piriform cortex and hippocampus following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2010, 518, 4612–4628. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; Frassoni, C.; Tassi, L.; Spreafico, R.; Garbelli, R. Aquaporin 4 expression in control and epileptic human cerebral cortex. Brain Res. 2011, 1367, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; Lee, T.S.; Thomas, M.J.; Amiry-Moghaddam, M.; Bjørnsen, L.P.; Spencer, D.D.; Agre, P.; Ottersen, O.P.; de Lanerolle, N.C. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Heuser, K.; Nagelhus, E.A.; Taubøll, E.; Indahl, U.; Berg, P.R.; Lien, S.; Nakken, S.; Gjerstad, L.; Ottersen, O.P. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 2010, 88, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Yao, X.; Verkman, A.S.; Manley, G.T. Increased seizure duration in mice lacking aquaporin-4 water channels. Acta Neurochir. Suppl. 2006, 96, 389–392. [Google Scholar] [PubMed]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Bazil, C.W. New molecular targets for antiepileptic drugs:alpha(2)delta, SV2A, and K(v)7/KCNQ/M potassium channels. Curr. Neurol. Neurosci. Rep. 2008, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Gee, N.S.; Brown, J.P.; Dissanayake, V.U.; Offord, J.; Thurlow, R.; Woodruff, G.N. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 1996, 271, 5768–5776. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, U.; Scherbaum, N. How addictive are gabapentin and pregabalin? A systematic review. Eur. Neuropsychopharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Calandre, E.P.; Rico-Villademoros, F.; Slim, M. Alpha(2)delta ligands, gabapentin, pregabalin and mirogabalin: A review of their clinical pharmacology and therapeutic use. Expert Rev. Neurother. 2016, 16, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Pulman, J.; Hutton, J.L.; Marson, A.G. Gabapentin add-on for drug-resistant partial epilepsy. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Costa, J.; Fareleira, F.; Ascenção, R.; Borges, M.; Sampaio, C.; Vaz-Carneiro, A. Clinical comparability of the new antiepileptic drugs in refractory partial epilepsy: A systematic review and meta-analysis. Epilepsia 2011, 52, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Shanthanna, H.; Gilron, I.; Rajarathinam, M.; AlAmri, R.; Kamath, S.; Thabane, L.; Devereaux, P.J.; Bhandari, M. Benefits and safety of gabapentinoids in chronic low back pain: A systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2017, 14, e1002369. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.S.; Freitas, M.F.; Rocha, I.R.; Chacur, M. Gabapentin decreases microglial cells and reverses bilateral hyperalgesia and allodynia in rats with chronic myositis. Eur. J. Pharmacol. 2017, 799, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Reda, H.M.; Zaitone, S.A.; Moustafa, Y.M. Effect of levetiracetam versus gabapentin on peripheral neuropathy and sciatic degeneration in streptozotocin-diabetic mice: Influence on spinal microglia and astrocytes. Eur. J. Pharmacol. 2016, 771, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Wodarski, R.; Clark, A.K.; Grist, J.; Marchand, F.; Malcangio, M. Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur. J. Pain 2009, 13, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.M.; de Brito, T.V.; de Aguiar Magalhães, D.; da Silva Santos, P.W.; Batista, J.A.; do Nascimento Dias, E.G.; de Barros Fernandes, H.; Damasceno, S.R.; Silva, R.O.; Aragão, K.S.; et al. Gabapentin, a synthetic analogue of gamma aminobutyric acid, reverses systemic acute inflammation and oxidative stress in mice. Inflammation 2014, 37, 1826–1836. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, H.; Badan, I.; Grecksch, G.; Walker, L.; Kessler, C.; Popa-Wagner, A. Kindling status in sprague-dawley rats induced by pentylenetetrazole: Involvement of a critical development period. Am. J. Pathol. 2003, 162, 1027–1034. [Google Scholar] [CrossRef]
- Buga, A.M.; Vintilescu, R.; Balseanu, A.T.; Pop, O.T.; Streba, C.; Toescu, E.; Popa-Wagner, A. Repeated PTZ treatment at 25-day intervals leads to a highly efficient accumulation of doublecortin in the dorsal hippocampus of rats. PLoS ONE 2012, 7, e39302. [Google Scholar] [CrossRef] [PubMed]
- Aviles-Reyes, R.X.; Angelo, M.F.; Villarreal, A.; Rios, H.; Lazarowski, A.; Ramos, A.J. Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: Implications for sleep apnea. J. Neurochem. 2010, 112, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Mothe, A.J.; Kulbatski, I.; van Bendegem, R.L.; Lee, L.; Kobayashi, E.; Keating, A.; Tator, C.H. Analysis of green fluorescent protein expression in transgenic rats for tracking transplanted neural stem/progenitor cells. J. Histochem. Cytochem. 2005, 53, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, A.; Rosciszewski, G.; Murta, V.; Cadena, V.; Usach, V.; Dodes-Traian, M.M.; Setton-Avruj, P.; Barbeito, L.H.; Ramos, A.J. Isolation and Characterization of Ischemia-Derived Astrocytes (IDAs) with Ability to Transactivate Quiescent Astrocytes. Front. Cell. Neurosci. 2016, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Murta, V.; Farías, M.I.; Pitossi, F.J.; Ferrari, C.C. Chronic systemic IL-1β exacerbates central neuroinflammation independently of the blood-brain barrier integrity. J. Neuroimmunol. 2015, 278, 30–43. [Google Scholar] [CrossRef] [PubMed]













© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, A.; Murta, V.; Auzmendi, J.; Ramos, A.J. Early Gabapentin Treatment during the Latency Period Increases Convulsive Threshold, Reduces Microglial Activation and Macrophage Infiltration in the Lithium-Pilocarpine Model of Epilepsy. Pharmaceuticals 2017, 10, 93. https://doi.org/10.3390/ph10040093
Rossi A, Murta V, Auzmendi J, Ramos AJ. Early Gabapentin Treatment during the Latency Period Increases Convulsive Threshold, Reduces Microglial Activation and Macrophage Infiltration in the Lithium-Pilocarpine Model of Epilepsy. Pharmaceuticals. 2017; 10(4):93. https://doi.org/10.3390/ph10040093
Chicago/Turabian StyleRossi, Alicia, Veronica Murta, Jerónimo Auzmendi, and Alberto Javier Ramos. 2017. "Early Gabapentin Treatment during the Latency Period Increases Convulsive Threshold, Reduces Microglial Activation and Macrophage Infiltration in the Lithium-Pilocarpine Model of Epilepsy" Pharmaceuticals 10, no. 4: 93. https://doi.org/10.3390/ph10040093
APA StyleRossi, A., Murta, V., Auzmendi, J., & Ramos, A. J. (2017). Early Gabapentin Treatment during the Latency Period Increases Convulsive Threshold, Reduces Microglial Activation and Macrophage Infiltration in the Lithium-Pilocarpine Model of Epilepsy. Pharmaceuticals, 10(4), 93. https://doi.org/10.3390/ph10040093