Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon?


{kind=link}
{kind=link}
Abstract
:1. The Disease
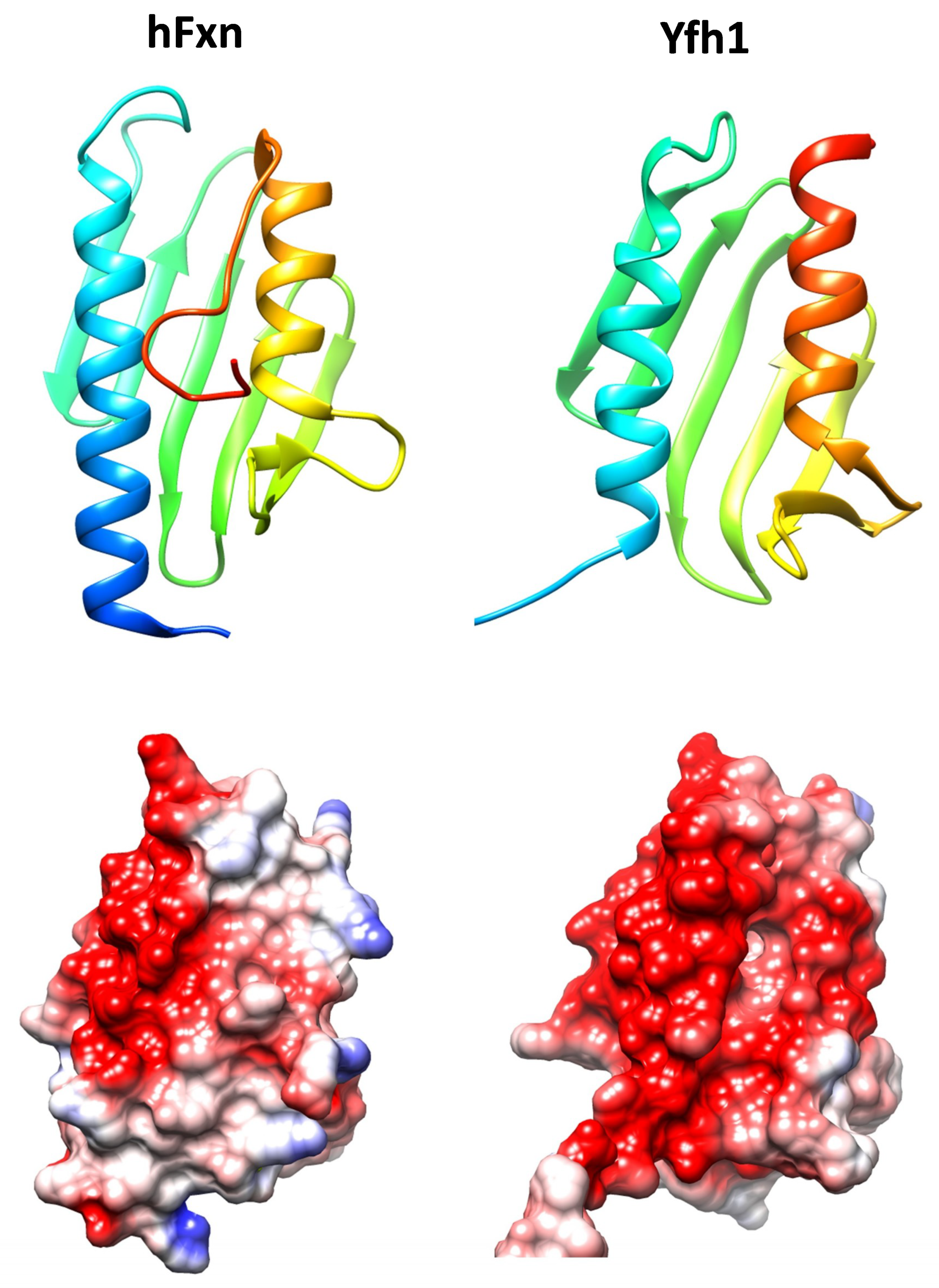
2. Frataxin, an Ancestral Conserved Protein
3. Frataxin Function
3.1. Frataxin, an Iron Binding and Storage Protein
3.2. Frataxin in the Biosynthesis of Iron Containing Proteins
3.2.1. Biosynthesis of Heme Groups
3.2.2. Biosynthesis of Iron-Sulfur Centers
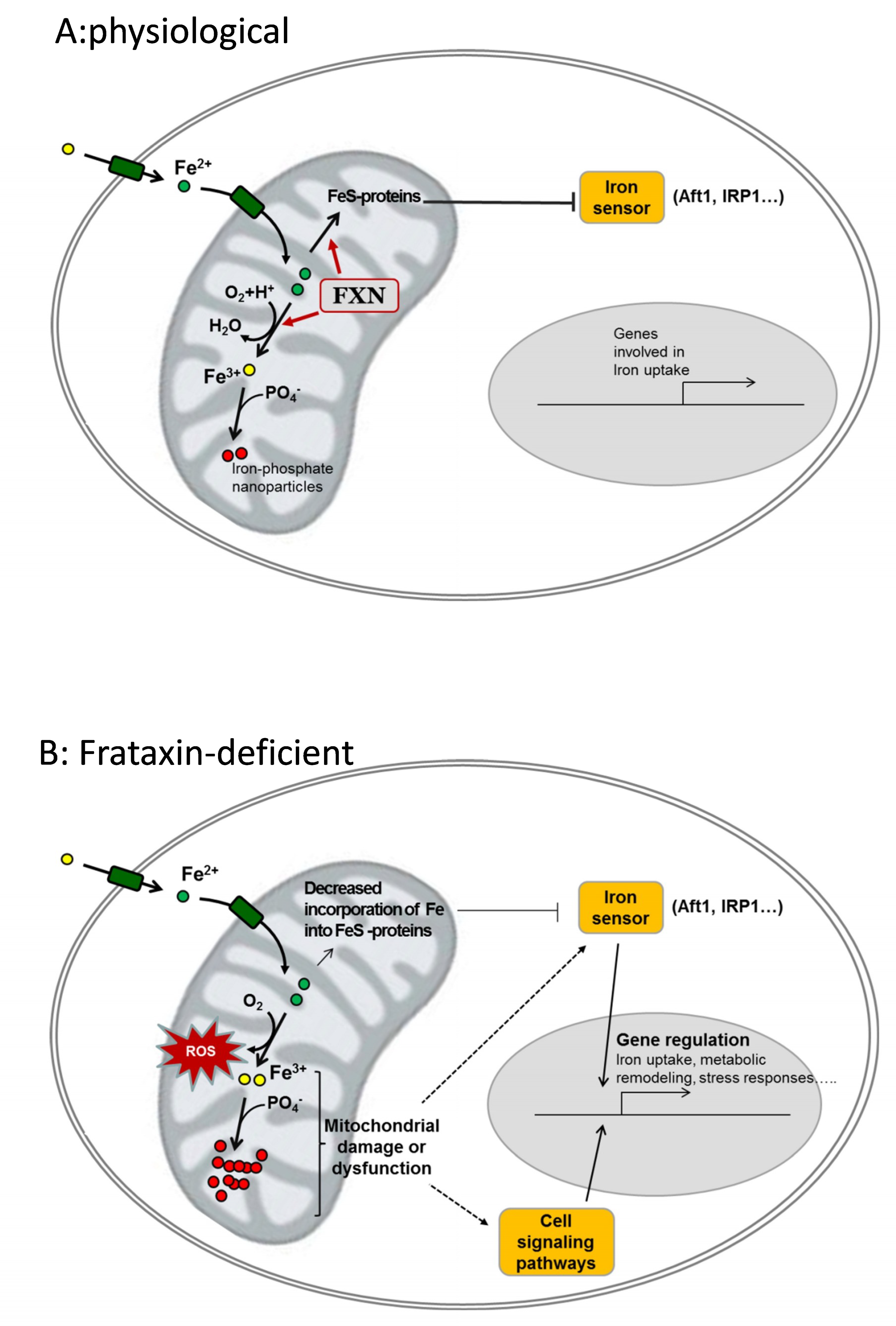
3.3. Control of Oxidative Stress and the Generation of Ros
4. Evidences of Iron Accumulation and Its Relation to Pathophysiology in FRDA
5. Targeting Iron as a Therapeutic Approach in FRDA
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Ashby, D.W.; Tweedy, P.S. Friedreich’s ataxia combined with diabetes mellitus in sisters. Br. Med. J. 1953, 1, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is Reduced in Friedreich Ataxia Patients and is Associated with Mitochondrial Membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filla, A.; De Michele, G.; Cavalcanti, F.; Pianese, L.; Monticelli, A.; Campanella, G.; Cocozza, S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996, 59, 554–560. [Google Scholar] [PubMed]
- Isnard, R.; Kalotka, H.; Dürr, A.; Cossée, M.; Schmitt, M.; Pousset, F.; Thomas, D.; Brice, A.; Koenig, M.; Komajda, M. Correlation between left ventricular hypertrophy and GAA trinucleotide repeat length in Friedreich’s ataxia. Circulation 1997, 95, 2247–2249. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, D.O.; Keats, B.; Vedanarayanan, V.; Subramony, S.H. Sequence variation in GAA repeat expansions may cause differential phenotype display in Friedreich’s ataxia. Mov. Disord. 2001, 16, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Evans-Galea, M.V.; Carrodus, N.; Rowley, S.M.; Corben, L.A.; Tai, G.; Saffery, R.; Galati, J.C.; Wong, N.C.; Craig, J.M.; Lynch, D.R.; et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012, 71, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Nair, M.; Politou, A.; Bayer, E.; Martin, S.; Temussi, P.; Pastore, A. The factors governing the thermal stability of frataxin orthologues: How to increase a protein’s stability. Biochemistry 2004, 43, 6511–6518. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Campuzano, V.; Koenig, M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum. Mol. Genet. 1998, 7, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branda, S.S.; Cavadini, P.; Adamec, J.; Kalousek, F.; Taroni, F.; Isaya, G. Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J. Biol. Chem. 1999, 274, 22763–22769. [Google Scholar] [CrossRef] [PubMed]
- Condò, I.; Ventura, N.; Malisan, F.; Rufini, A.; Tomassini, B.; Testi, R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007, 16, 1534–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmucker, S.; Argentini, M.; Carelle-Calmels, N.; Martelli, A.; Puccio, H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008, 17, 3521–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gakh, O.; Bedekovics, T.; Duncan, S.F.; Smith, D.Y.; Berkholz, D.S.; Isaya, G. Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron-sulfur cluster assembly. J. Biol. Chem. 2010, 285, 38486–38501. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, P.; Adamec, J.; Taroni, F.; Gakh, O.; Isaya, G. Two-step Processing of Human Frataxin by Mitochondrial Processing Peptidase. J. Biol. Chem. 2000, 275, 41469–41475. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Y.; Dai, X.; Marelja, Z.; Zhou, D.; Mo, R.; Al-Mahdawi, S.; Pook, M.A.; Leimkühler, S.; Rouault, T.A.; et al. Novel frataxin isoforms may contribute to the pathological mechanism of Friedreich ataxia. PLoS ONE 2012, 7, e47847. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Iron−Sulfur Cluster Biosynthesis. Characterization of Frataxin as an Iron Donor for Assembly of [2Fe−2S] Clusters in ISU-Type Proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Gentry, L.E.; Thacker, M.A.; Doughty, R.; Timkovich, R.; Busenlehner, L.S. His86 from the N-Terminus of Frataxin Coordinates Iron and Is Required for Fe-S Cluster Synthesis. Biochemistry 2013, 52, 6085–6096. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. J. Inorg. Biochem. 2018, 183, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Adamec, J.; Rusnak, F.; Owen, W.G.; Naylor, S.; Benson, L.M.; Gacy, M.; Isaya, G. Iron-dependent self-assembly of recombinant yeast frataxin: Implications for Friedreich ataxia. Am. J. Hum. Genet. 2000, 67, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schagerlof, U.; Elmlund, H.; Gakh, O.; Nordlund, G.; Hebert, H.; Lindahl, M.; Isaya, G.; Al-Karadaghi, S. Structural basis of the iron storage function of frataxin from single-particle reconstruction of the iron-loaded oligomer. Biochemistry 2008, 47, 4948–4954. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Isaya, G.; O’Neill, H.A.; Santambrogio, P.; Cozzi, A.; Arosio, P.; Levi, S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum. Mol. Genet. 2004, 13, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Trifuoggi, M.; Politou, A.S.; Martin, S.; Pastore, A. A structural approach to understanding the iron-binding properties of phylogenetically different frataxins. Hum. Mol. Genet. 2002, 11, 1865–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–D658. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, E.C.; Fekry, M.; Wiemann, M.; Söderberg, C.A.; Bernfur, K.; Gakh, O.; Rasmussen, M.; Højrup, P.; Emanuelsson, C.; Isaya, G.; et al. Iron-induced oligomerization of human FXN81-210 and bacterial CyaY frataxin and the effect of iron chelators. PLoS ONE 2017, 12, e0188937. [Google Scholar] [CrossRef] [PubMed]
- Seguin, A.; Sutak, R.; Bulteau, A.L.; Garcia-Serres, R.; Oddou, J.L.; Lefevre, S.; Santos, R.; Dancis, A.; Camadro, J.; Latour, J.; et al. Evidence that yeast frataxin is not an iron storage protein in vivo. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, B.; Bistrich, U.; Schranzhofer, M.; Sarsero, J.P.; Rauen, U.; Scheiber-mojdehkar, B.; de Groot, H.; Ioannou, P.; Petrat, F. Friedreich’s Ataxia, No Changes in Mitochondrial Labile Iron in Human Lymphoblasts and Fibroblasts A Decrease in Antioxidative Capacity? J. Biol. Chem. 2004, 280, 6701–6708. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.A.; Moore, M.J. Labile Low-Molecular-Mass Metal Complexes in Mitochondria: Trials and Tribulations of a Burgeoning Field. Biochemistry 2016, 55, 4140–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber-Morales, J.; Holmes-Hampton, G.P.; Miao, R.; Guo, Y.; Münck, E.; Lindahl, P.A. Biophysical characterization of iron in mitochondria isolated from respiring and fermenting yeast. Biochemistry 2010, 49, 5436–5444. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Gakh, O.; O’Neill, H.; Mangravita, A.; Nichol, H.; Ferreira, G.C.; Isaya, G. Yeast frataxin sequentially chaperones and stores iron by coupling protein assembly with iron oxidation. J. Biol. Chem. 2003, 278, 31340–31351. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Alam, S.L.; Proteasa, S.V.; Zhang, Y.; Lesuisse, E.; Dancis, A.; Stemmler, T.L. Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry 2004, 43, 16254–16262. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, C.G.; Gillam, M.E.; Ahlgren, E.C.; Hunter, G.A.; Gakh, O.; Isaya, G.; Ferreira, G.C.; Al-Karadaghi, S. The Structure of the Complex between Yeast Frataxin and Ferrochelatase: Characterization and pre-Steady State Reaction of Ferrous Iron Delivery and Heme Synthesis. J. Biol. Chem. 2016, 291, 11887–11898. [Google Scholar] [CrossRef] [PubMed]
- Steinkellner, H.; Singh, H.N.; Muckenthaler, M.U.; Goldenberg, H.; Moganty, R.R.; Scheiber-Mojdehkar, B.; Sturm, B. No changes in heme synthesis in human Friedreich’s ataxia erythroid progenitor cells. Gene 2017, 621, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Cortopassi, G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 2007, 457, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cermeno, A.; Obis, E.; Belli, G.; Cabiscol, E.; Ros, J.; Tamarit, J. Frataxin Depletion in Yeast Triggers Up-regulation of Iron Transport Systems before Affecting Iron-Sulfur Enzyme Activities. J. Biol. Chem. 2010, 285, 41653–41664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Cermeno, A.; Alsina, D.; Cabiscol, E.; Tamarit, J.; Ros, J. Metabolic remodeling in frataxin-deficient yeast is mediated by Cth2 and Adr1. Biochim. Biophys. Acta 2013, 1833, 3326–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donze, M.; Reutenauer, L.; Puccio, H. Mammalian Frataxin: An Essential Function for Cellular Viability through an Interaction with a Preformed ISCU/NFS1/ISD11 Iron-Sulfur Assembly Complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Craig, E.A. Binding of yeast frataxin to the scaffold for Fe-S cluster biogenesis. J. Biol. Chem. 2008, 283, 12674–12679. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autréaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed]
- Llorens, J.; Navarro, J.; Martínez-Sebastián, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.; Moltó, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Obis, È.; Irazusta, V.; Sanchís, D.; Ros, J.; Tamarit, J. Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free Radic. Biol. Med. 2014, 73, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, N.; Ayte, J.; Hidalgo, E. Cells lacking pfh1, a fission yeast homolog of Mammalian frataxin protein, display constitutive activation of the iron starvation response. J. Biol. Chem. 2012, 287, 43042–43051. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, J.; Obis, È.; Ros, J. Oxidative stress and altered lipid metabolism in Friedreich ataxia. Free Radic. Biol. Med. 2016, 100, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Bayot, A.; Santos, R.; Camadro, J.M.; Rustin, P. Friedreich’s ataxia: The vicious circle hypothesis revisited. BMC Med. 2011, 9, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Irazusta, V.; Obis, E.; Moreno-Cermeño, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Yeast frataxin mutants display decreased superoxide dismutase activity crucial to promote protein oxidative damage. Free Radic. Biol. Med. 2010, 48, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron Regulatory Protein 1 Sustains Mitochondrial Iron Loading and Function in Frataxin Deficiency. Cell Metab. 2015, 21, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Dancis, A.; Gareil, M.; Montagne, J.J.; Camadro, J.M.; Lesuisse, E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 2007, 42, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J.; Koonin, E.; Musco, G.; Pastore, A.; Bork, P. Friedreich’s ataxia protein: Phylogenetic evidence for mitochondrial dysfunction. Trends Neurosci. 1996, 19, 465–468. [Google Scholar] [CrossRef]
- Anderson, P.R.; Kirby, K.; Orr, W.C.; Hilliker, A.J.; Phillips, J.P. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc. Natl. Acad. Sci. USA 2008, 105, 611–616. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.A.; Gakh, O.; Park, S.; Cui, J.; Mooney, S.M.; Sampson, M.; Ferreira, G.C.; Isaya, G. Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry 2005, 44, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Gakh, O.; Park, S.; Liu, G.; Macomber, L.; Imlay, J.A.; Ferreira, G.C.; Isaya, G. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum. Mol. Genet. 2006, 15, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cabo, P.; Vazquez-Manrique, R.P.; Garcia-Gimeno, M.A.; Sanz, P.; Palau, F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum. Mol. Genet. 2005, 14, 2091–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dolle, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Lodi, R.; Rajagopalan, B.; Blamire, A.M.; Cooper, J.M.; Davies, C.H.; Bradley, J.L.; Styles, P.; Schapira, A. Cardiac energetics are abnormal in Friedreich ataxia patients in the absence of cardiac dysfunction and hypertrophy: An in vivo 31P magnetic resonance spectroscopy study. Cardiovasc. Res. 2001, 52, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.S.; Khdour, O.; Hecht, S.M. Does oxidative stress contribute to the pathology of Friedreich’s ataxia? A radical question. FASEB J. 2010, 24, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Lesuisse, E.; Santos, R.; Matzanke, B.F.; Knight, S.A.B.; Camadro, J.M.; Dancis, A. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 2003, 12, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsina, D.; Ros, J.; Tamarit, J. Nitric oxide prevents Aft1 activation and metabolic remodeling in frataxin-deficient yeast. Redox Biol. 2018, 14, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.Y.; Li, T.; Li, Z.; Duraine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. Elife 2016, 5, e16043. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Rahmanto, Y.S.; Sutak, R.; Xu, X.; Becker, E.M.; Mikhael, M.R.; Ponka, P.; Richardson, D.R. The MCK mouse heart model of Friedreich’s ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl. Acad. Sci. USA 2008, 105, 9757–9762. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.L.; Qian, J.; Santambrogio, P.; Levi, S.; Koeppen, A.H. Relation of Cytosolic Iron Excess to Cardiomyopathy of Friedreich’s Ataxia. Am. J. Cardiol. 2012, 110, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Rahmanto, Y.S.; Huang, M.L.H.; Saletta, F.; Lok, H.C.; Gutierrez, L.; Lázaro, F.J.; Fleming, A.J.; St. Pierre, T.G.; Mikhael, M.R.; et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. USA 2012, 109, 20590–20595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Ho, T.S.Y.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. Elife 2016, 5, e20732. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The dentate nucleus in Friedreich’s ataxia: The role of iron-responsive proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Simon, D.; Monassier, L.; Criqui-Filipe, P.; Gansmuller, A.; Rustin, P.; Koenig, M.; Puccio, H. Idebenone delays the onset of cardiac functional alteration without correction of Fe-S enzymes deficit in a mouse model for Friedreich ataxia. Hum. Mol. Genet. 2004, 13, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehling, O.; Elsässer, H.P.; Brückel, B.; Mühlenhoff, U.; Lill, R. Iron-sulfur protein maturation in human cells: Evidence for a function of frataxin. Hum. Mol. Genet. 2004, 13, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Condò, I.; Malisan, F.; Guccini, I.; Serio, D.; Rufini, A.; Testi, R. Molecular control of the cytosolic aconitase/IRP1 switch by extramitochondrial frataxin. Hum. Mol. Genet. 2010, 19, 1221–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Seguin, A.; Santos, R.; Pain, D.; Dancis, A.; Camadro, J.M.; Lesuisse, E. Co-precipitation of phosphate and iron limits mitochondrial phosphate availability in Saccharomyces cerevisiae lacking the yeast frataxin homologue (YFH1). J. Biol. Chem. 2011, 286, 6071–6079. [Google Scholar] [CrossRef] [PubMed]
- Purroy, R.; Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial pore opening and loss of Ca2+ exchanger NCLX levels occur after frataxin depletion. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Irazusta, V.; Cabiscol, E.; Reverter-Branchat, G.; Ros, J.; Tamarit, J. Manganese is the link between frataxin and iron-sulfur deficiency in the yeast model of Friedreich ataxia. J. Biol. Chem. 2006, 281, 12227–12232. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Calap-Quintana, P.; Llorens, J.V.; Al-Ramahi, I.; Gutiérrez, L.; Martínez-Sebastián, M.J.; Botas, J.; Moltó, M.D. Metal Homeostasis Regulators Suppress FRDA Phenotypes in a Drosophila Model of the Disease. PLoS ONE 2016, 11, e0159209. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Kuntzsch, E.C.; Bjork, S.T.; Ramirez, R.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich ataxia: Metal dysmetabolism in dorsal root ganglia. Acta Neuropathol. Commun. 2013, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Tasheva, S.; Obis, E.; Tamarit, J.; Ros, J. Apoptotic cell death and altered calcium homeostasis caused by frataxin depletion in dorsal root ganglia neurons can be prevented by BH4 domain of Bcl-xL protein. Hum. Mol. Genet. 2013, 23, 1829–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.H.L.; Camadro, J.M.; Santos, R.; Lesuisse, E.; Chahine, J.M.; Ha-Duong, N.T. Mechanisms of iron and copper-frataxin interactions. Metallomics 2017, 9, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Tai, G.; Corben, L.A.; Yiu, E.M.; Milne, S.C.; Delatycki, M.B. Progress in the treatment of Friedreich ataxia. Neurol. Neurochir. Pol. 2018, 52, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Britti, E.; Delaspre, F.; Feldman, A.; Osborne, M.; Greif, H.; Tamarit, J.; Ros Salvador, J. Frataxin-deficient neurons and mice models of Friedreich ataxia are improved by TAT-MTScs-FXN treatment. J. Cell. Mol. Med. 2017, 22, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Manning, H.; Breuer, W.; Melamed-Book, N.; Lu, C.; Cortopassi, G.; Munnich, A.; Cabantchik, Z. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood 2008, 112, 5219–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandolfo, M.; Hausmann, L. Deferiprone for the treatment of Friedreich’s ataxia. J. Neurochem. 2013, 126, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Elincx-Benizri, S.; Glik, A.; Merkel, D.; Arad, M.; Freimark, D.; Kozlova, E.; Cabantchik, I.; Hassin-Baer, S. Clinical Experience With Deferiprone Treatment for Friedreich Ataxia. J. Child Neurol. 2016, 31, 1036–1040. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsina, D.; Purroy, R.; Ros, J.; Tamarit, J. Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon? Pharmaceuticals 2018, 11, 89. https://doi.org/10.3390/ph11030089
Alsina D, Purroy R, Ros J, Tamarit J. Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon? Pharmaceuticals. 2018; 11(3):89. https://doi.org/10.3390/ph11030089
Chicago/Turabian StyleAlsina, David, Rosa Purroy, Joaquim Ros, and Jordi Tamarit. 2018. "Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon?" Pharmaceuticals 11, no. 3: 89. https://doi.org/10.3390/ph11030089
APA StyleAlsina, D., Purroy, R., Ros, J., & Tamarit, J. (2018). Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon? Pharmaceuticals, 11(3), 89. https://doi.org/10.3390/ph11030089