Iron Regulation: Macrophages in Control


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
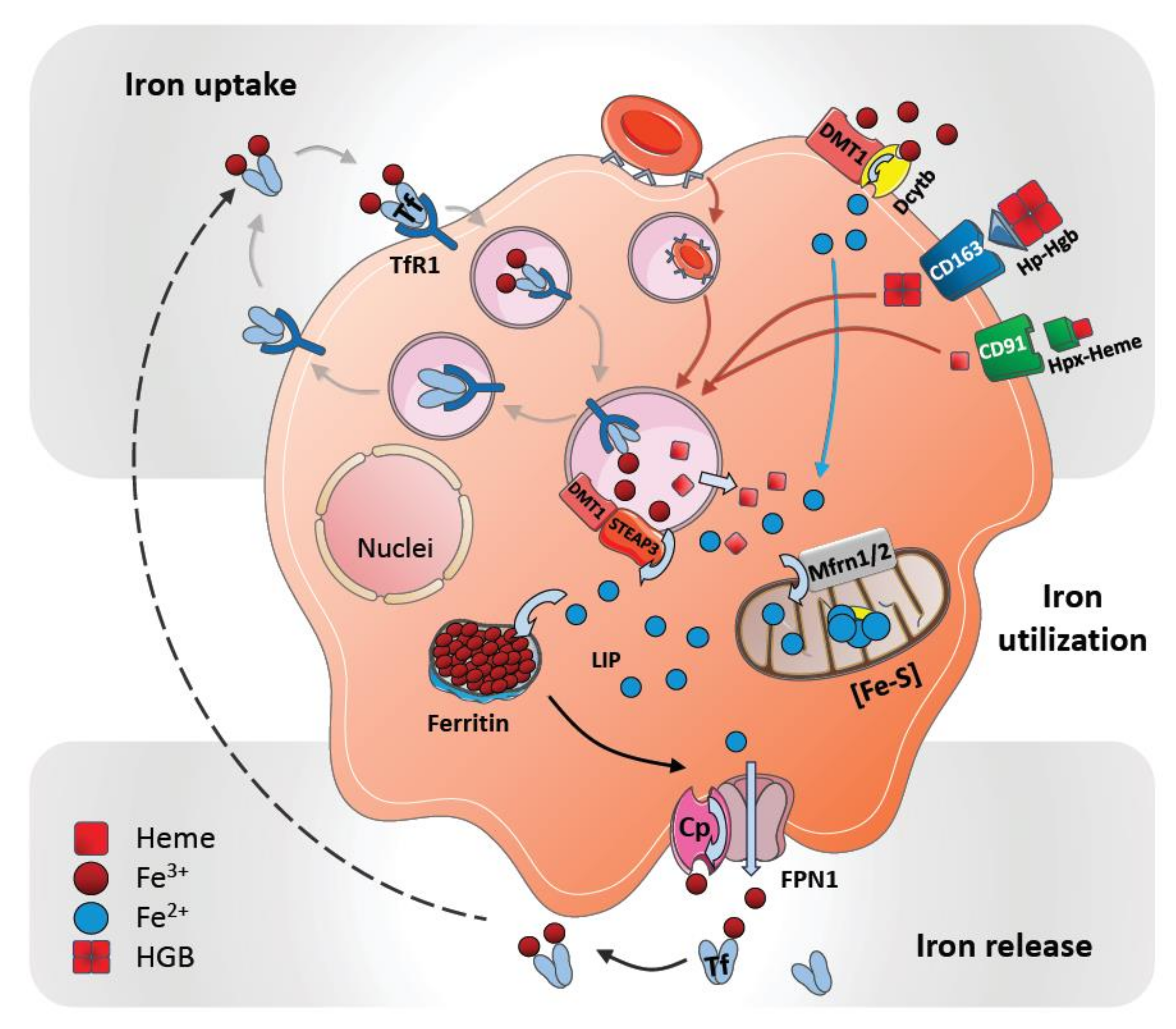
2. Cellular Uptake and Metabolism of Iron in Macrophages
3. Systemic Iron Metabolism
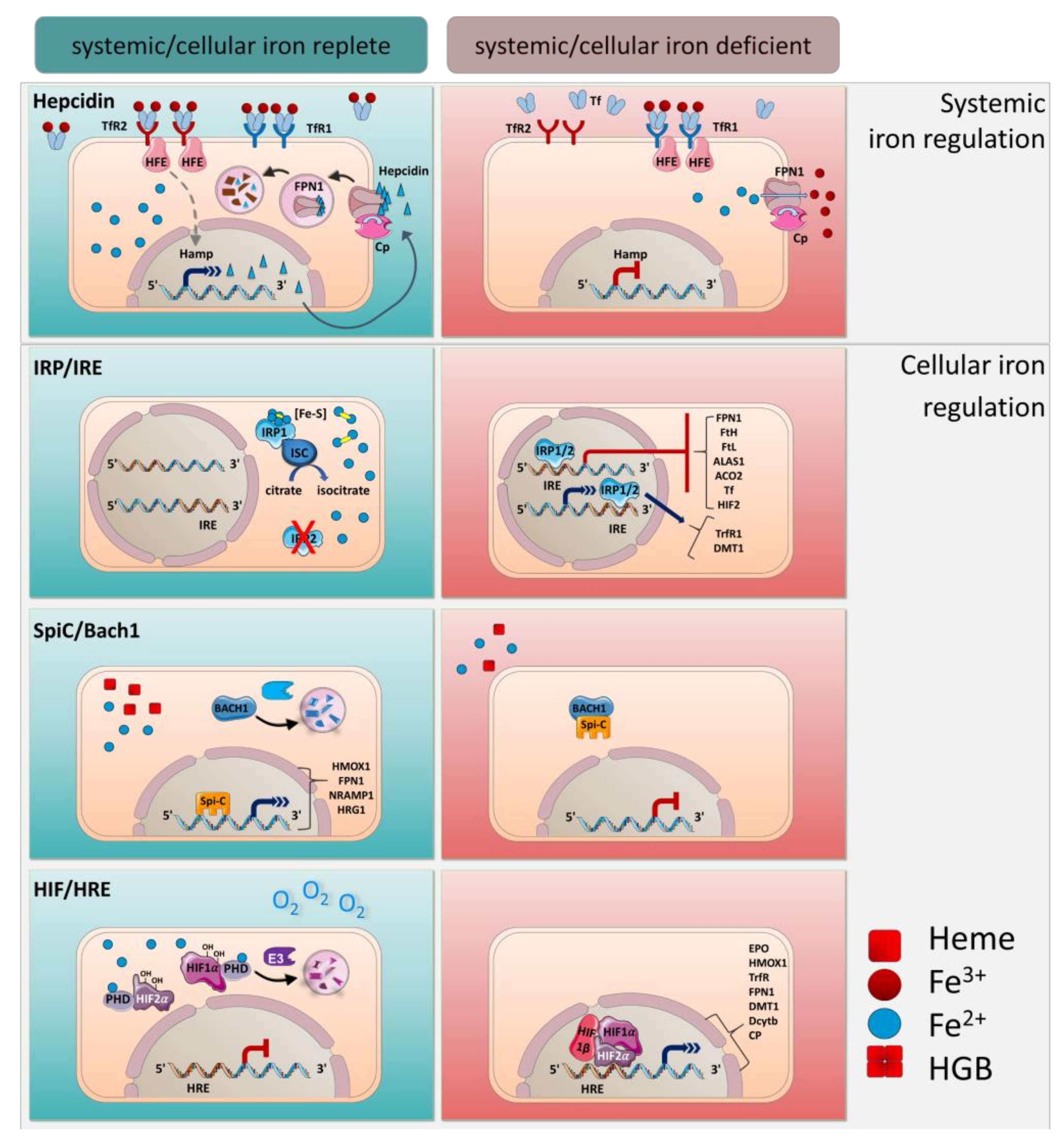
4. Systemic Iron Regulation by Hepcidin
5. Cellular Regulation of Iron by IRE/IRPs
6. Transcriptional Regulation by Spi-C and HIF
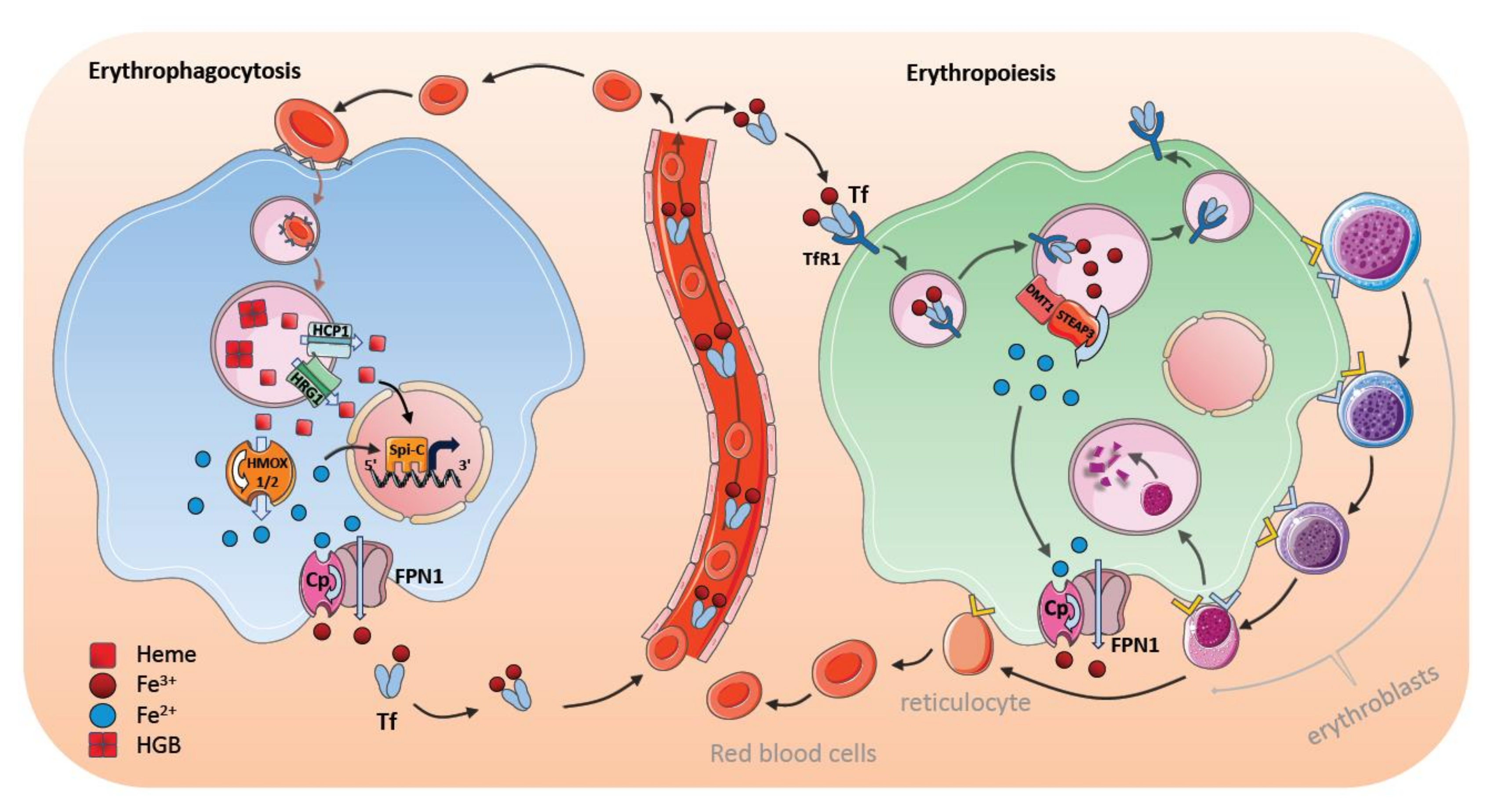
7. Steady-State Erythrophagocytosis by Macrophages in the Spleen
8. Stress-Induced Erythrophagocytosis and Iron Metabolism
9. Bone Marrow Macrophages and Erythroblastic Islands
10. Regulation of Iron Transfer by Central Nurse Macrophages
11. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACO2 | aconitase 2 |
| ALAS1 | delta-aminolevulinate synthase 1 |
| BACH1 | BTB domain and CNC homolog 1 |
| BM | bone marrow |
| BMP | Bone morphogenetic protein |
| Cp | ceruloplasmin |
| DCT1 | divalent cation transporter 1 |
| DcytB | duodenal cytochrome B |
| DMT1 | divalent metal transporter 1 |
| E3 | E3 ubiquitin ligase |
| Epo | erythroid factor erythropoietin |
| FLVCR1 | Feline leukemia virus group C cellular receptor 1 |
| FPN1 | ferroportin 1 |
| FtMt | Mitochondrial ferritin |
| FtH | heavy chain H-ferritin |
| FtL | light chain L-ferritin |
| GDF15 | growth differentiation factor 15 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| Hamp | hepcidin antimicrobial peptide |
| HEPH | hephaestin |
| HCP1 | heme-carrier protein 1 |
| HFE | homeostatic iron regulator |
| Hgb | hemoglobin |
| HIF1α/HIF2α | hypoxia-inducible factor 1α/2α |
| HJV | hemojuvelin |
| HMOX1 | heme oxygenase 1 |
| Hp | haptoglobin |
| Hpx | hemopexin |
| HRE | hypoxia-responsive element |
| HRG1 | heme responsive gene 1 protein |
| ILGF1 | insulin-like growth factor 1 |
| IFNγ | interferon gamma |
| IRE | iron-regulatory element |
| IRP1/IRP2 | iron-regulatory protein1/2 |
| ISC | iron-sulfur cluster |
| KC | Kupffer cell |
| LIP | Labile iron pool |
| Mfrn1/2 | Mitoferrin 1/2 |
| MerTK | MER proto-oncogene tyrosine kinase |
| NTBI | non-transferrin-bound iron |
| NRAMP1 | natural resistance-associated macrophage protein 1 |
| PHDs | oxygen prolyl hydroxylases |
| PS | phosphatidylserine |
| PV | polycythemia vera |
| RBC | red blood cell |
| RPM | red pulp macrophages |
| SIRPα | signal-regulatory protein alpha |
| Spi-C | Spi-1/PU.1 related transcription factor |
| SR-AI | scavenger receptor type A member I |
| STAT3 | activator of transcription 3 |
| STEAP3 | six-transmembrane epithelial antigen of the prostate 3 |
| Tf | transferrin |
| TfR1/2 | transferrin receptor 1/2 |
| TNFα | tumor necrosis factor alpha |
| TGF-β | transforming growth factor beta |
| TWSG1 | twisted gastrulation BMP signaling modulator 1 |
| VCAM1 | vascular cell adhesion protein 1 |
| UTR | untranslated region |
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Hamza, I. Macrophages and iron metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [PubMed]
- Fabiano, A.; Brilli, E.; Mattii, L.; Testai, L.; Moscato, S.; Citi, V.; Tarantino, G.; Zambito, Y. Ex vivo and in vivo study of sucrosomial® iron intestinal absorption and bioavailability. Int. J. Mol. Sci. 2018, 19, 2722. [Google Scholar] [CrossRef] [PubMed]
- Orr, J.S.; Kennedy, A.; Anderson-Baucum, E.K.; Webb, C.D.; Fordahl, S.C.; Erikson, K.M.; Zhang, Y.; Etzerodt, A.; Moestrup, S.K.; Hasty, A.H. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes 2014, 63, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kim, M.S.; Han, S.N. Diet-induced obesity leads to decreased hepatic iron storage in mice. Nutr. Res. 2011, 31, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Festa, M.; Ricciardelli, G.; Mele, G.; Pietropaolo, C.; Ruffo, A.; Colonna, A. Overexpression of H ferritin and up-regulation of iron regulatory protein genes during differentiation of 3T3-L1 pre-adipocytes. J. Biol. Chem. 2000, 275, 36708–36712. [Google Scholar] [CrossRef]
- Fleming, M.D.; Andrews, N.C. Mammalian iron transport: An unexpected link between metal homeostasis and host defense. J. Lab. Clin. Med. 1998, 132, 464–468. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping iron”-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017, 469, 397–418. [Google Scholar] [CrossRef]
- Gruenheid, S.; Pinner, E.; Desjardins, M.; Gros, P. Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 1997, 185, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.R.; Gros, P. Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane. Blood 2003, 102, 1884–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soe-Lin, S.; Sheftel, A.D.; Wasyluk, B.; Ponka, P. Nramp1 equips macrophages for efficient iron recycling. Exp. Hematol. 2008, 36, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Nakai, Y.; Ueda, S.; Kamigaso, S.; Ohta, K.; Hatakeyama, M.; Hayashi, Y.; Otagiri, M.; Yuasa, H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G660–G668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Vallelian, F.; Imhof, A.; Schoedon, G.; Schaer, D.J. Heme carrier protein (HCP-1) spatially interacts with the CD163 hemoglobin uptake pathway and is a target of inflammatory macrophage activation. J. Leukoc. Biol. 2008, 83, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Zhang, J.; Chambers, I.; Yun, S.; Phillips, J.; Krause, M.; Hamza, I. Hrg1 promotes heme-iron recycling during hemolysis in the zebrafish kidney. PLoS Genet. 2018, 14, e1007665. [Google Scholar] [CrossRef]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS ONE 2012, 7, e42199. [Google Scholar] [CrossRef]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Seaman, W.E.; Di, X.; Wang, W.; Willingham, M.; Torti, F.M.; Torti, S.V. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE 2011, 6, e23800. [Google Scholar] [CrossRef] [PubMed]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Springer, A.; Weisheit, D.; Petrat, F.; Korth, H.-G.; de Groot, H.; Sustmann, R. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chembiochem 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Yewdall, S.J.; Harrison, P.M.; Santambrogio, P.; Cozzi, A.; Rovida, E.; Albertini, A.; Arosio, P. Evidence of H- and L-chains have co-operative roles in the iron-uptake mechanism of human ferritin. Biochem. J. 1992, 288 Pt 2, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Delaby, C.; Pilard, N.; Hetet, G.; Driss, F.; Grandchamp, B.; Beaumont, C.; Canonne-Hergaux, F. A physiological model to study iron recycling in macrophages. Exp. Cell Res. 2005, 310, 43–53. [Google Scholar] [CrossRef]
- Ferreira, C.; Santambrogio, P.; Martin, M.E.; Andrieu, V.; Feldmann, G.; Hénin, D.; Beaumont, C. H ferritin knockout mice: A model of hyperferritinemia in the absence of iron overload. Blood 2001, 98, 525–532. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Zhang, A.-S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Montosi, G.; Donovan, A.; Totaro, A.; Garuti, C.; Pignatti, E.; Cassanelli, S.; Trenor, C.C.; Gasparini, P.; Andrews, N.C.; Pietrangelo, A. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J. Clin. Investig. 2001, 108, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Njajou, O.T.; Vaessen, N.; Joosse, M.; Berghuis, B.; van Dongen, J.W.; Breuning, M.H.; Snijders, P.J.; Rutten, W.P.; Sandkuijl, L.A.; Oostra, B.A.; et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet. 2001, 28, 213–214. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Dini, L.; Carbonaro, M.; Musci, G.; Calabrese, L. The interaction of ceruloplasmin with Kupffer cells. Eur. J. Cell Biol. 1990, 52, 207–212. [Google Scholar]
- De Domenico, I.; Ward, D.M.; di Patti, M.C.B.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef] [Green Version]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [Green Version]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar] [CrossRef] [PubMed]
- Kennard, M.L.; Richardson, D.R.; Gabathuler, R.; Ponka, P.; Jefferies, W.A. A novel iron uptake mechanism mediated by GPI-anchored human p97. EMBO J. 1995, 14, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Frazer, D.M.; Wilkins, S.J.; Becker, E.M.; Murphy, T.L.; Vulpe, C.D.; McKie, A.T.; Anderson, G.J. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut 2003, 52, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupic, F.; Fruchon, S.; Bensaid, M.; Loreal, O.; Brissot, P.; Borot, N.; Roth, M.P.; Coppin, H. Duodenal mRNA expression of iron related genes in response to iron loading and iron deficiency in four strains of mice. Gut 2002, 51, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. The anemia of “haemoglobin-deficit” (hbd/hbd) mice is caused by a defect in transferrin cycling. Exp. Hematol. 2006, 34, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, J.N.; Conrad, M.E.; Moore, E.G.; Latour, L.F. Iron absorption and cellular transport: The mobilferrin/paraferritin paradigm. Semin. Hematol. 1998, 35, 13–26. [Google Scholar] [PubMed]
- Zhang, D.-L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Yeh, K.-Y.; Yeh, M.; Glass, J. Interactions between ferroportin and hephaestin in rat enterocytes are reduced after iron ingestion. Gastroenterology 2011, 141, 292–299.e1. [Google Scholar] [CrossRef]
- Yeh, K.-Y.; Yeh, M.; Mims, L.; Glass, J. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G55–G65. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005, 2, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, T.A.M.; Mauk, A.G.; MacGillivray, R.T.A. Recombinant expression and functional characterization of human hephaestin: A multicopper oxidase with ferroxidase activity. Biochemistry 2005, 44, 14725–14731. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Preza, G.C.; Jung, C.-L.; Kaplan, J.; Waring, A.J.; Ganz, T. The N-terminus of hepcidin is essential for its interaction with ferroportin: Structure-function study. Blood 2006, 107, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Kulaksiz, H.; Gehrke, S.G.; Janetzko, A.; Rost, D.; Bruckner, T.; Kallinowski, B.; Stremmel, W. Pro-hepcidin: Expression and cell specific localisation in the liver and its regulation in hereditary haemochromatosis, chronic renal insufficiency, and renal anaemia. Gut 2004, 53, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.-J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Valore, E.V.; Nemeth, E.; Goodnough, J.B.; Gabayan, V.; Ganz, T. Iron transferrin regulates hepcidin synthesis in primary hepatocyte culture through hemojuvelin and BMP2/4. Blood 2007, 110, 2182–2189. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA 2002, 99, 4596–4601. [Google Scholar] [CrossRef] [Green Version]
- Altamura, S.; Kessler, R.; Gröne, H.-J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003, 33, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Hepcidin: A putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8160–8162. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.B.; Chen, J.; Murchison, N.; Green, F.A.; Enns, C.A. Transferrin receptor 2: Evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol. Biol. Cell 2007, 18, 743–754. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Viatte, L.; Lou, D.-Q.; Bennoun, M.; Beaumont, C.; Kahn, A.; Andrews, N.C.; Vaulont, S. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat. Genet. 2003, 34, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.-S.; Enns, C.A. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009, 9, 217–227. [Google Scholar] [CrossRef]
- Ajioka, R.S.; Levy, J.E.; Andrews, N.C.; Kushner, J.P. Regulation of iron absorption in Hfe mutant mice. Blood 2002, 100, 1465–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef]
- Ramey, G.; Deschemin, J.-C.; Vaulont, S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009, 94, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H.; Fleming, R.E.; Gui, D.; Moon, S.Y.; Saitoh, T.; O’Kelly, J.; Umehara, Y.; Wano, Y.; Said, J.W.; Koeffler, H.P. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 2005, 105, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.-P.; Townsend, A.R.M.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef]
- Soares, M.P.; Weiss, G. The Iron age of host-microbe interactions. EMBO Rep. 2015, 16, 1482–1500. [Google Scholar] [CrossRef] [Green Version]
- Pasricha, S.-R.; McHugh, K.; Drakesmith, H. Regulation of hepcidin by erythropoiesis: The story so far. Annu. Rev. Nutr. 2016, 36, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.P.; Ribeiro, S.; Pontes, H.; Thowfeequ, S.; Tosh, D.; Carvalho, F.; Porto, G. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood 2008, 111, 5727–5733. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.-H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.C.; Wang, R.-H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.-J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Iron regulatory elements (IREs): A family of mRNA non-coding sequences. Biochem. J. 1994, 304 Pt 1, 1–11. [Google Scholar] [CrossRef]
- Hentze, M.W.; Caughman, S.W.; Rouault, T.A.; Barriocanal, J.G.; Dancis, A.; Harford, J.B.; Klausner, R.D. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987, 238, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: Regulatory RNA sequences that control mRNA levels and translation. Science 1988, 240, 924–928. [Google Scholar] [CrossRef]
- Dandekar, T.; Hentze, M.W. Finding the hairpin in the haystack: Searching for RNA motifs. Trends Genet. 1995, 11, 45–50. [Google Scholar] [CrossRef]
- Evstatiev, R.; Gasche, C. Iron sensing and signalling. Gut 2012, 61, 933–952. [Google Scholar] [CrossRef]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fillebeen, C.; Chen, G.; Biederbick, A.; Lill, R.; Pantopoulos, K. Iron-dependent degradation of apo-IRP1 by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2007, 27, 2423–2430. [Google Scholar] [CrossRef]
- Wang, J.; Chen, G.; Lee, J.; Pantopoulos, K. Iron-dependent degradation of IRP2 requires its C-terminal region and IRP structural integrity. BMC Mol. Biol. 2008, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Allerson, C.R.; Polycarpou-Schwarz, M.; Rofts, A.; Rogers, J.T.; Kishi, F.; Hentze, M.W.; Rouault, T.A.; Andrews, N.C.; Hediger, M.A. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001, 509, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Galy, B.; Ferring, D.; Minana, B.; Bell, O.; Janser, H.G.; Muckenthaler, M.; Schümann, K.; Hentze, M.W. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005, 106, 2580–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.-I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef]
- Ferring-Appel, D.; Hentze, M.W.; Galy, B. Cell-autonomous and systemic context-dependent functions of iron regulatory protein 2 in mammalian iron metabolism. Blood 2009, 113, 679–687. [Google Scholar] [CrossRef]
- Haldar, M.; Kohyama, M.; So, A.Y.-L.; Kc, W.; Wu, X.; Briseño, C.G.; Satpathy, A.T.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef]
- Kohyama, M.; Ise, W.; Edelson, B.T.; Wilker, P.R.; Hildner, K.; Mejia, C.; Frazier, W.A.; Murphy, T.L.; Murphy, K.M. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 2009, 457, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, D.; Uede, T.; Tamura, T. Functions and development of red pulp macrophages. Microbiol. Immunol. 2015, 59, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [PubMed]
- Warnatz, H.-J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M.; et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011, 286, 23521–23532. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Schimanski, L.M.; Ormerod, E.; Merryweather-Clarke, A.T.; Viprakasit, V.; Edwards, J.P.; Sweetland, E.; Bastin, J.M.; Cowley, D.; Chinthammitr, Y.; et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 2005, 106, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappalardi, M.B.; McNulty, D.E.; Martin, J.D.; Fisher, K.E.; Jiang, Y.; Burns, M.C.; Zhao, H.; Ho, T.; Sweitzer, S.; Schwartz, B.; et al. Biochemical characterization of human HIF hydroxylases using HIF protein substrates that contain all three hydroxylation sites. Biochem. J. 2011, 436, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, N.; Hentze, M.W. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: Implications for regulation and cellular function. Proc. Natl. Acad. Sci. USA 2002, 99, 12345–12350. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.-Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Fiorito, V.; Marro, S.; Silengo, L.; Altruda, F.; Tolosano, E. Cell-specific regulation of Ferroportin transcription following experimentally-induced acute anemia in mice. Blood Cells Mol. Dis. 2013, 50, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrogiannaki, M.; Matak, P.; Mathieu, J.R.R.; Delga, S.; Mayeux, P.; Vaulont, S.; Peyssonnaux, C. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica 2012, 97, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Nathan, D.G.; Ginsburg, D.; Look, A.T.; Fisher, D.E.; Lux, S. Nathan and Oski’s Hematology of Infancy and Childhood E-Book; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Ulyanova, T.; Padilla, S.M.; Papayannopoulou, T. Stage specific functional roles of integrins in erythropoiesis. Exp. Hematol. 2014, 2, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Bratosin, D.; Mazurier, J.; Tissier, J.P.; Estaquier, J.; Huart, J.J.; Ameisen, J.C.; Aminoff, D.; Montreuil, J. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie 1998, 80, 173–195. [Google Scholar] [CrossRef]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Beaumont, C.; Delaby, C. Recycling iron in normal and pathological states. Semin. Hematol. 2009, 46, 328–338. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Ghosh, M.C.; Ollivierre, W.; Weitzel, R.P.; Eckhaus, M.A.; Tisdale, J.F.; Yachie, A.; Rouault, T.A. Wild-type macrophages reverse disease in heme oxygenase 1-deficient mice. Blood 2014, 124, 1522–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.S.; Holderried, T.A.W.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabriek, B.O.; van Bruggen, R.; Deng, D.M.; Ligtenberg, A.J.M.; Nazmi, K.; Schornagel, K.; Vloet, R.P.M.; Dijkstra, C.D.; van den Berg, T.K. The macrophage scavenger receptor CD163 functions as an innate immune sensor for bacteria. Blood 2009, 113, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Graversen, J.H.; Madsen, M.; Moestrup, S.K. CD163: A signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int. J. Biochem. Cell Biol. 2002, 34, 309–314. [Google Scholar] [CrossRef]
- Eid, R.; Zhou, D.R.; Arab, N.T.T.; Boucher, E.; Young, P.G.; Mandato, C.A.; Greenwood, M.T. Heterologous expression of anti-apoptotic human 14-3-3β/α enhances iron-mediated programmed cell death in yeast. PLoS ONE 2017, 12, e0184151. [Google Scholar] [CrossRef]
- Martins, R.; Maier, J.; Gorki, A.-D.; Huber, K.V.M.; Sharif, O.; Starkl, P.; Saluzzo, S.; Quattrone, F.; Gawish, R.; Lakovits, K.; et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol. 2016, 17, 1361–1372. [Google Scholar] [CrossRef]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessis, M. Erythroblastic island, functional unity of bone marrow. Rev. Hematol. 1958, 13, 8–11. [Google Scholar] [PubMed]
- Mohandas, N.; Prenant, M. Three-dimensional model of bone marrow. Blood 1978, 51, 633–643. [Google Scholar] [PubMed]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the cradle to the grave: The role of macrophages in erythropoiesis and erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef]
- Sadahira, Y.; Yoshino, T.; Monobe, Y. Very late activation antigen 4-vascular cell adhesion molecule 1 interaction is involved in the formation of erythroblastic islands. J. Exp. Med. 1995, 181, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zermati, Y.; Fichelson, S.; Valensi, F.; Freyssinier, J.M.; Rouyer-Fessard, P.; Cramer, E.; Guichard, J.; Varet, B.; Hermine, O. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp. Hematol. 2000, 28, 885–894. [Google Scholar] [CrossRef]
- Toda, S.; Segawa, K.; Nagata, S. MerTK-mediated engulfment of pyrenocytes by central macrophages in erythroblastic islands. Blood 2014. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Iglesias, A.; Potocnik, A.J.; Hartmann, U.; Fässler, R. Impaired migration but not differentiation of haematopoietic stem cells in the absence of β1 integrins. Nature 1996, 380, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Heideveld, E.; van den Akker, E. Digesting the role of bone marrow macrophages on hematopoiesis. Immunobiology 2017, 222, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Casu, C.; Gardenghi, S.; Breda, L.; Crielaard, B.J.; Guy, E.; Marongiu, M.F.; Gupta, R.; Levine, R.L.; Abdel-Wahab, O.; et al. Macrophages support pathological erythropoiesis in polycythemia vera and β-thalassemia. Nat. Med. 2013, 19, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacchini, L.; Gammella, E.; De Ponti, C.; Recalcati, S.; Cairo, G. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J. Biol. Chem. 2008, 283, 20674–20686. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T.; Simpson, R.J.; Dawson, S.; Bouriez-Jones, T.; Crockford, T.L.; Lepherd, M.; Latunde-Dada, G.O.; Robinson, H.; Raja, K.B.; Campagna, D.R.; et al. Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood 2009, 113, 1805–1808. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Song, Y.; Zhang, Z.; Li, D.; Zhu, H.; Liang, R.; Gu, Y.; Pang, Y.; Qi, J.; Wu, H.; et al. Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice. J. Cereb. Blood Flow Metab. 2016, 37, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, C.A.; Deuel, J.W.; Schildknecht, D.; Mahmoudi, L.; Garcia-Rubio, I.; Owczarek, C.; Schauer, S.; Kissner, R.; Banerjee, U.; Palmer, A.F.; et al. Haptoglobin Preserves Vascular Nitric Oxide Signaling during Hemolysis. Am. J. Respir. Crit. Care Med. 2016, 193, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Leimberg, M.J.; Prus, E.; Konijn, A.M.; Fibach, E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J. Cell Biochem. 2008, 103, 1211–1218. [Google Scholar] [CrossRef]
- Leimberg, J.M.; Konijn, A.M.; Fibach, E. Developing human erythroid cells grown in transferrin-free medium utilize iron originating from extracellular ferritin. Am. J. Hematol. 2003, 73, 211–212. [Google Scholar] [CrossRef] [Green Version]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. https://doi.org/10.3390/ph11040137
Sukhbaatar N, Weichhart T. Iron Regulation: Macrophages in Control. Pharmaceuticals. 2018; 11(4):137. https://doi.org/10.3390/ph11040137
Chicago/Turabian StyleSukhbaatar, Nyamdelger, and Thomas Weichhart. 2018. "Iron Regulation: Macrophages in Control" Pharmaceuticals 11, no. 4: 137. https://doi.org/10.3390/ph11040137
APA StyleSukhbaatar, N., & Weichhart, T. (2018). Iron Regulation: Macrophages in Control. Pharmaceuticals, 11(4), 137. https://doi.org/10.3390/ph11040137