BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease



Abstract
:1. Introduction
1.1. Alzheimer’s Disease–Epidemiology
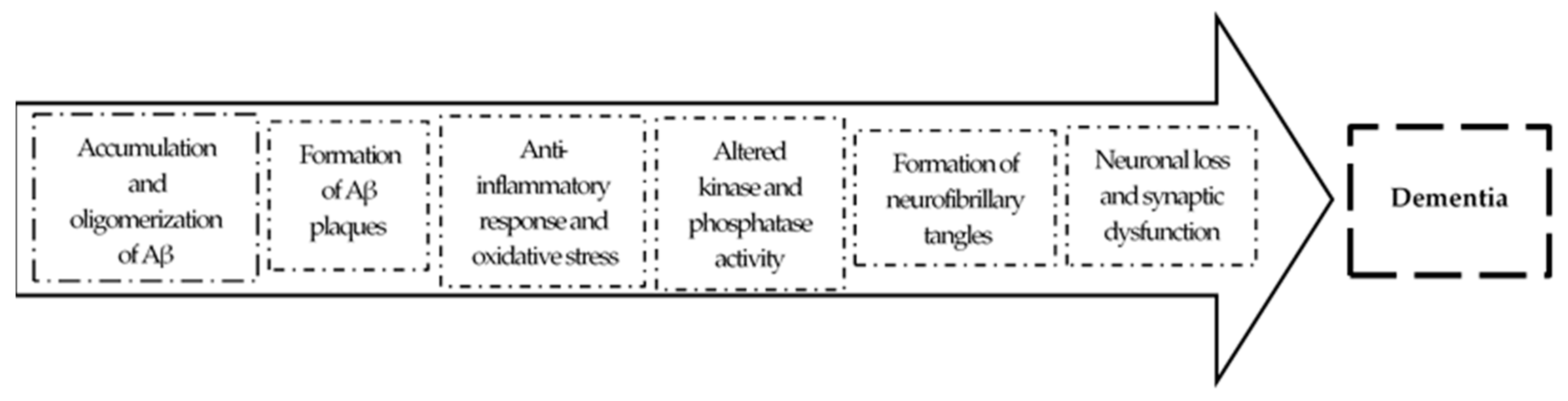
1.2. Pathology
1.3. Current Pharmacology and Drug Development
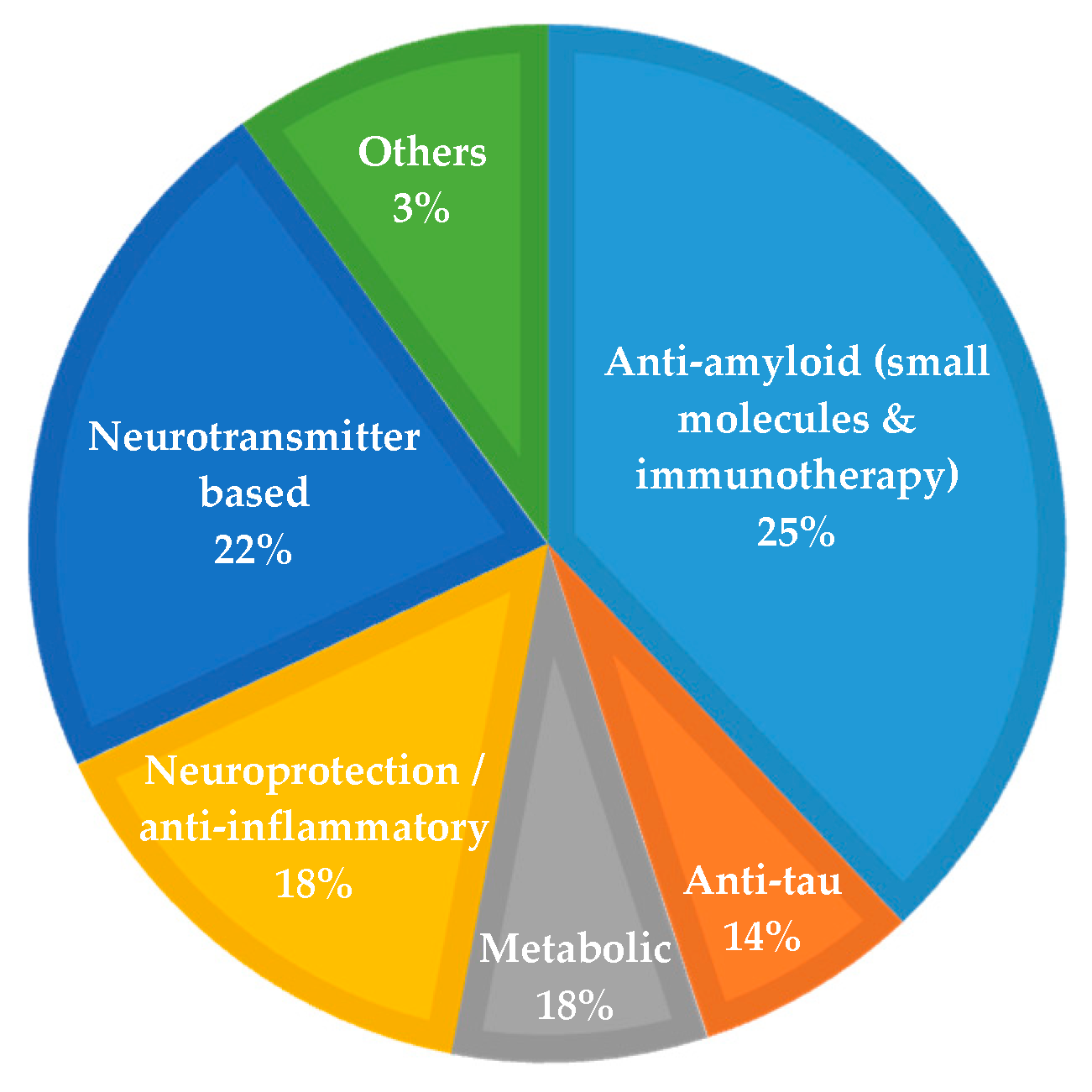
1.4. Disease-Modifying Therapies and Drug Development
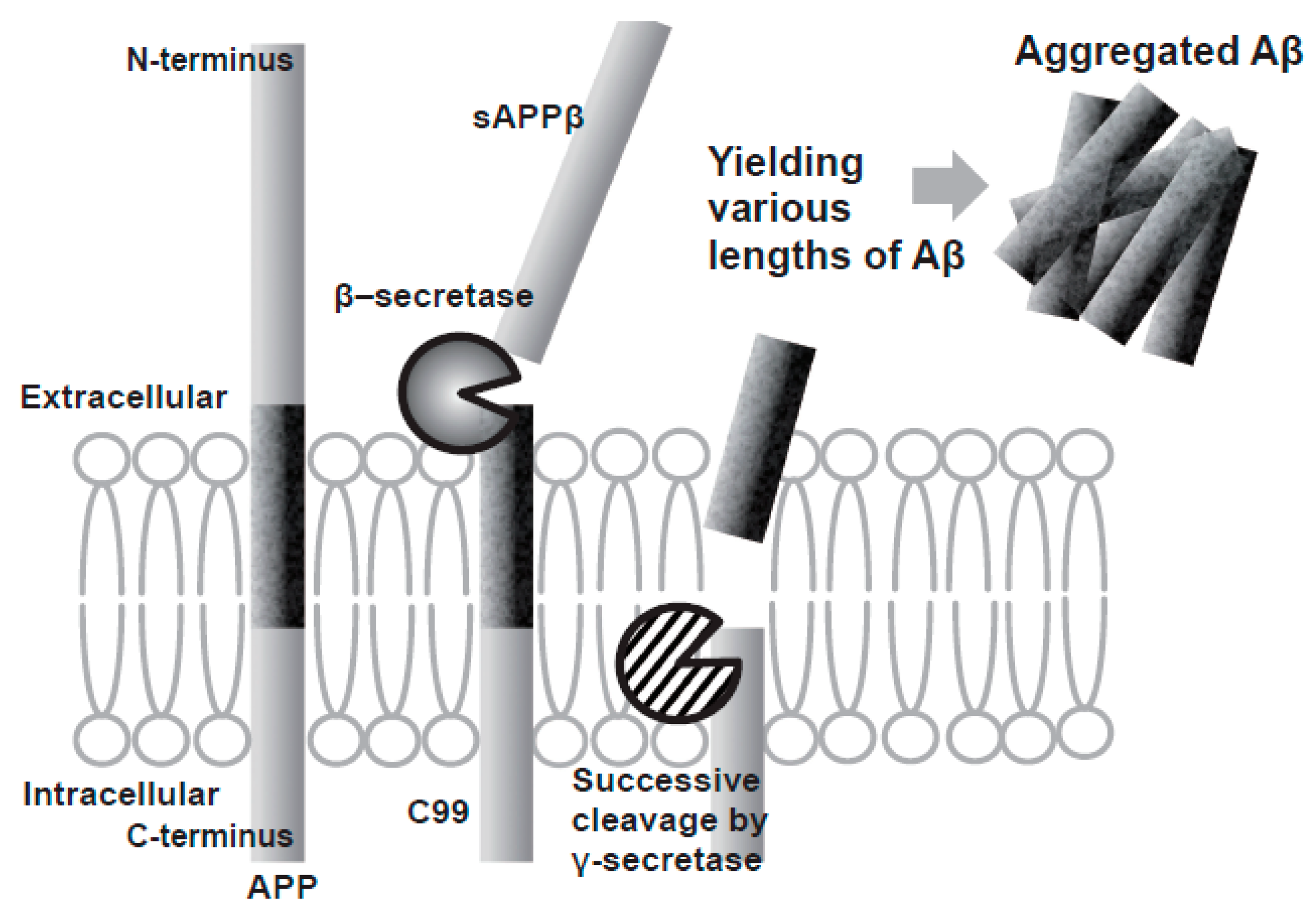
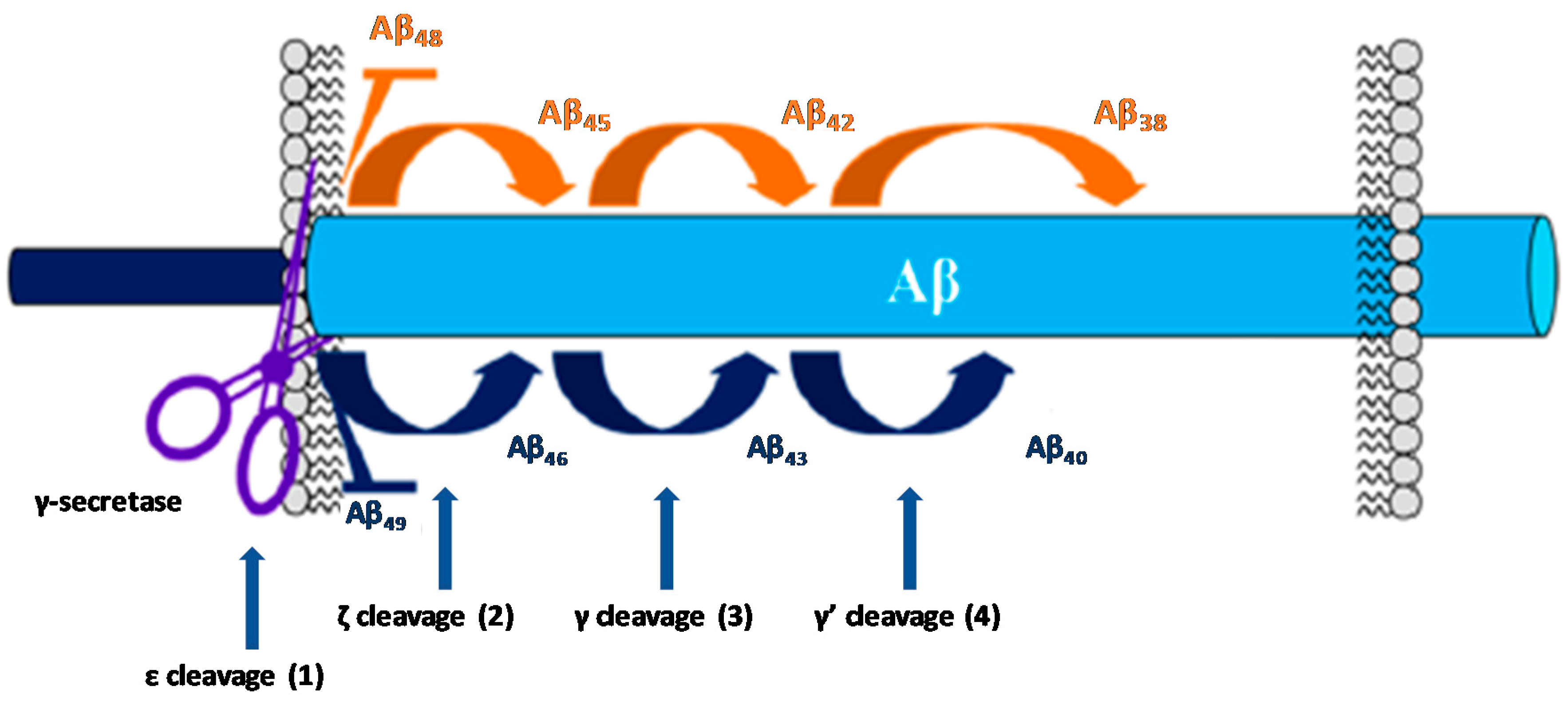
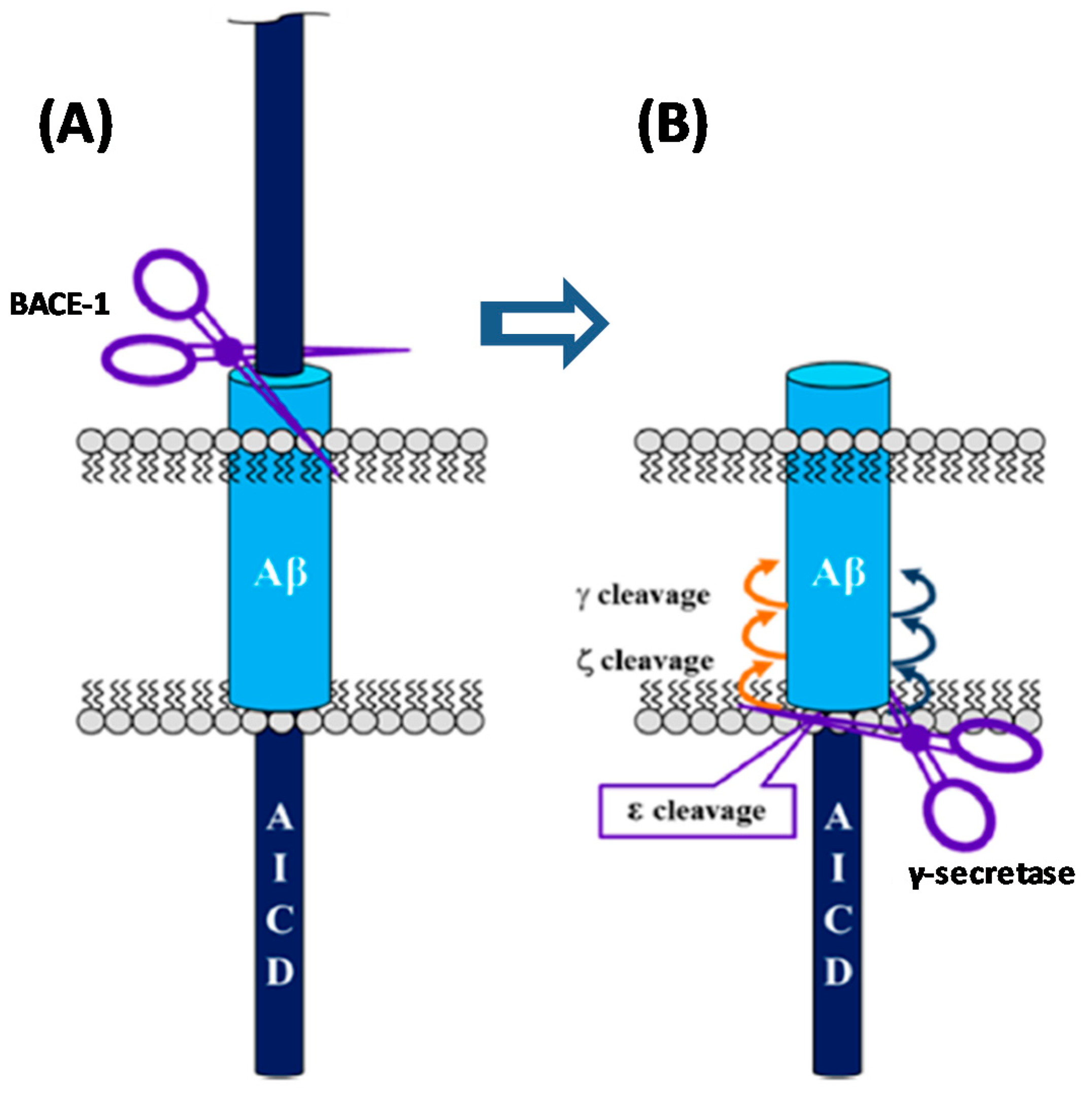
2. The Amyloid Hypothesis of AD
3. Amyloid Targeting Strategies
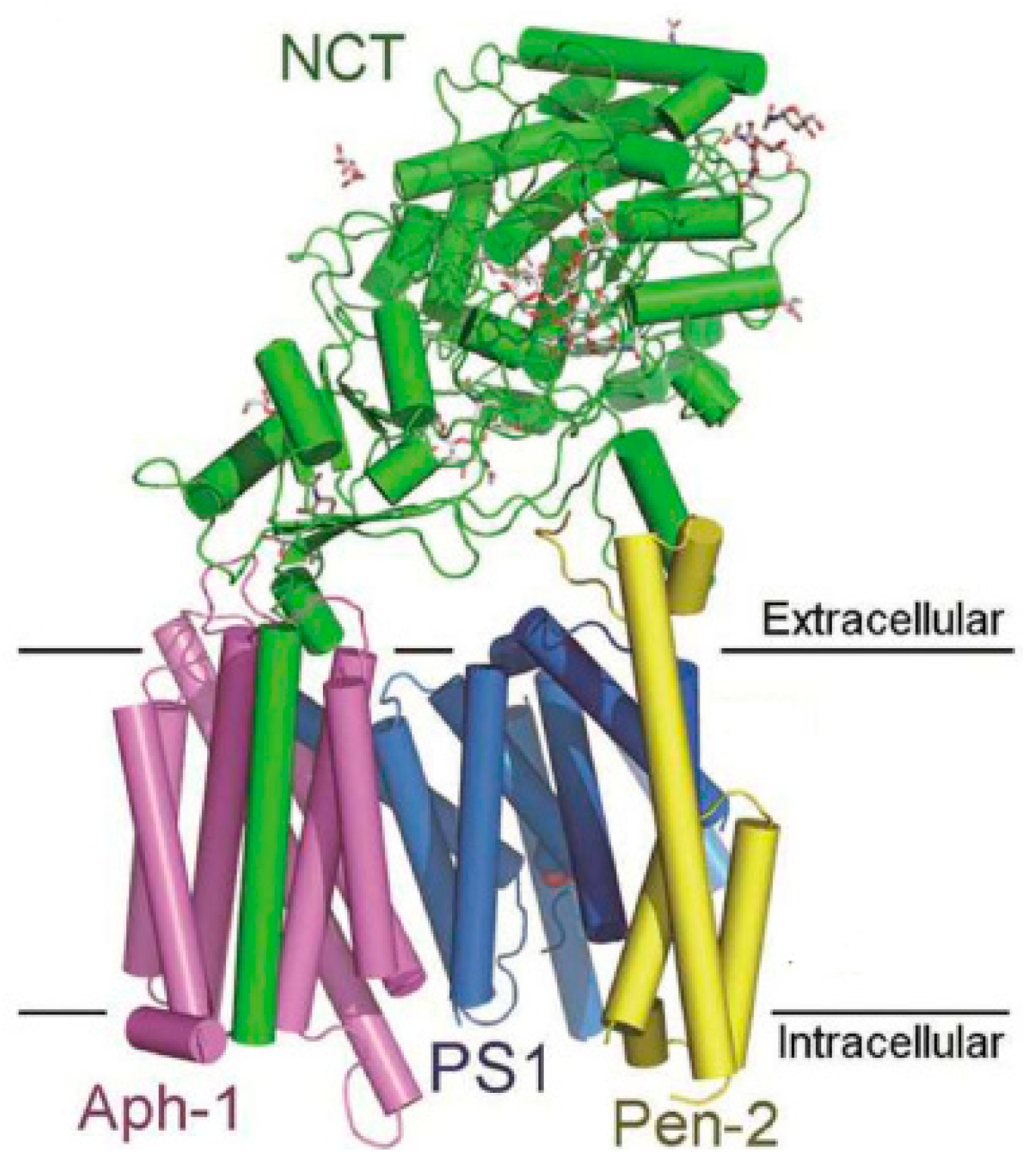
3.1. Gamma Secretase Inhibitors and Modulators
3.1.1. γ-Secretase Inhibitors
3.1.2. Gamma secretase Modulators
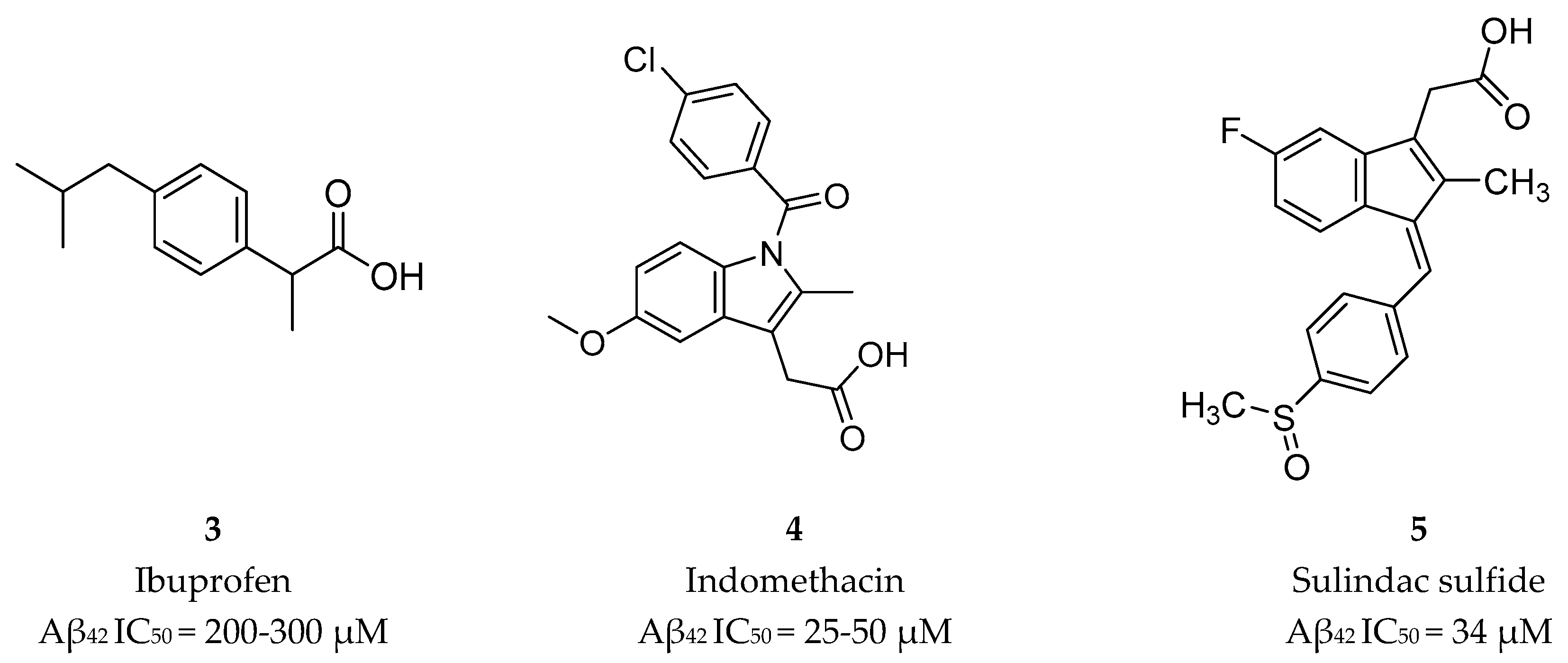
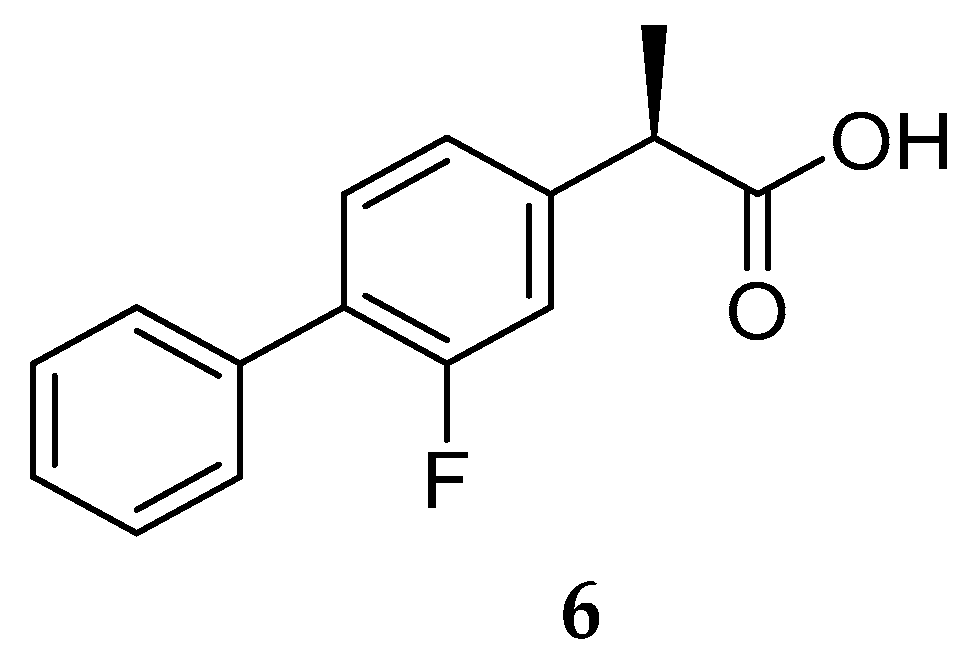
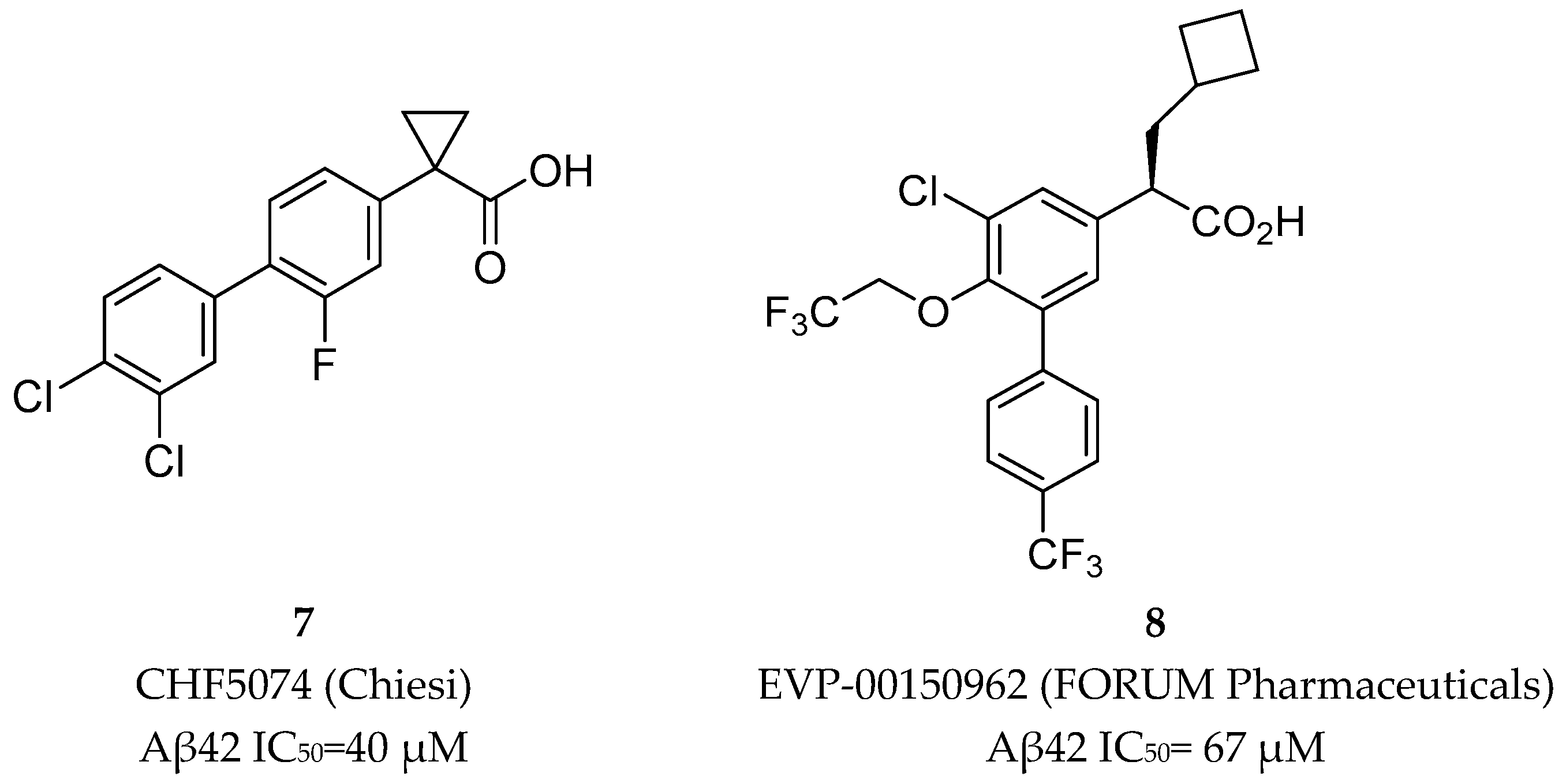
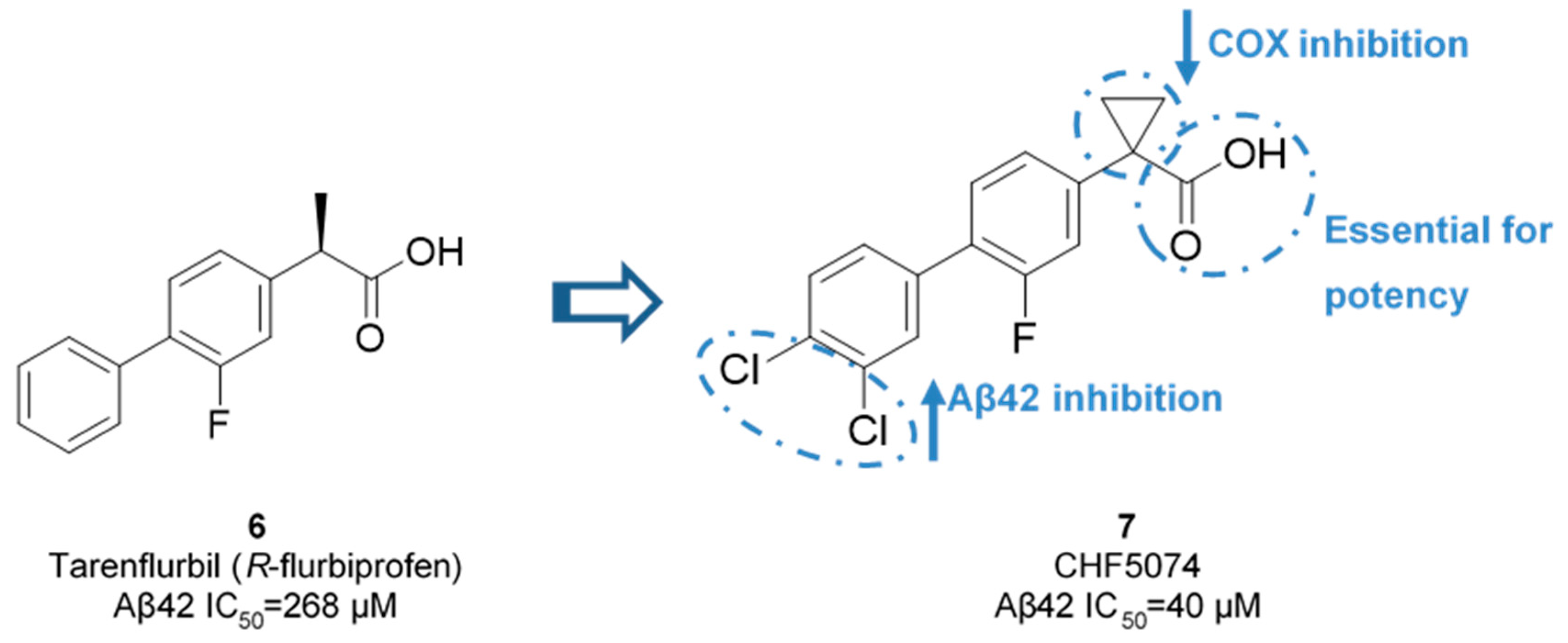
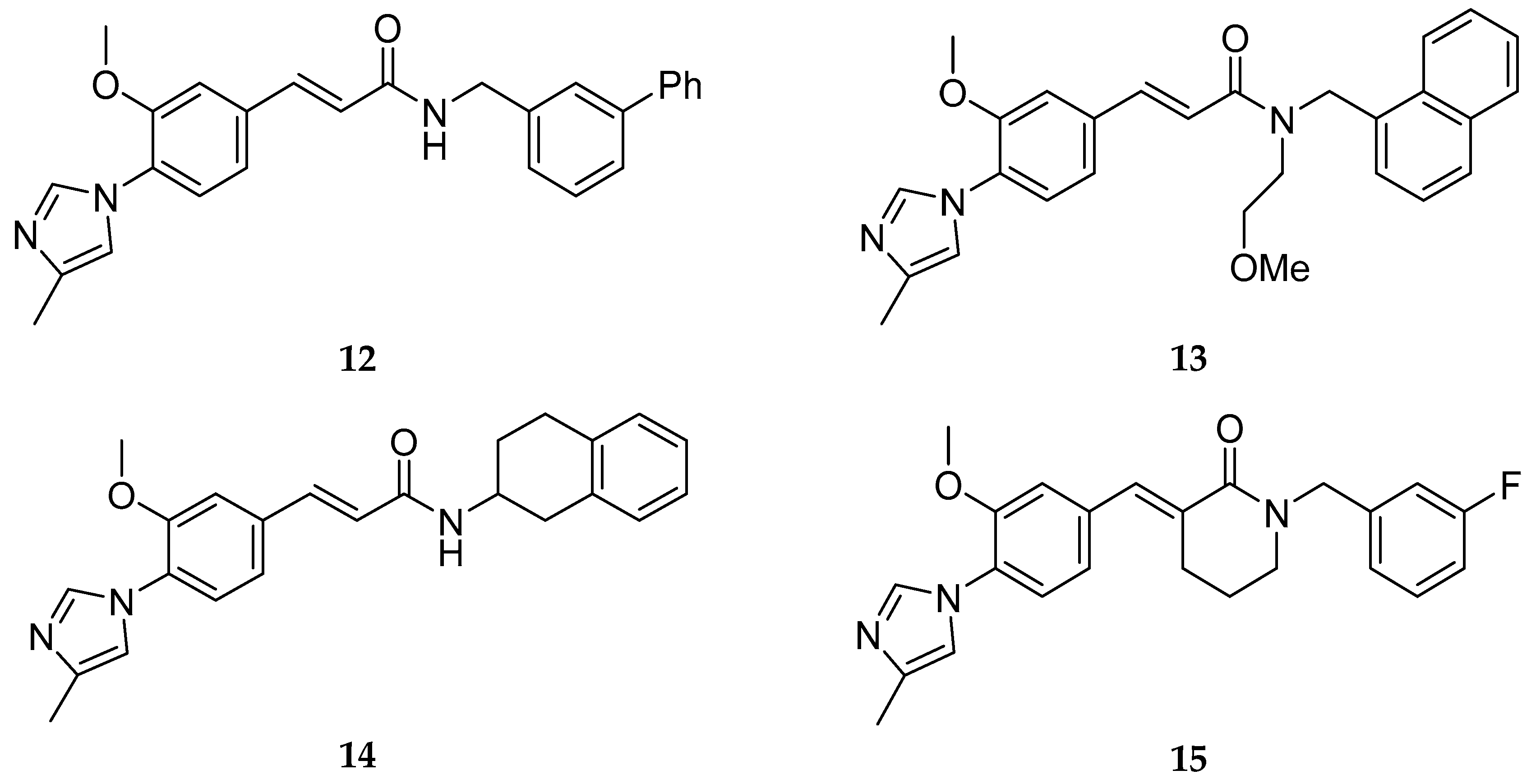
Nonsteroidal Anti-inflammatory Drug Derived GSM’s
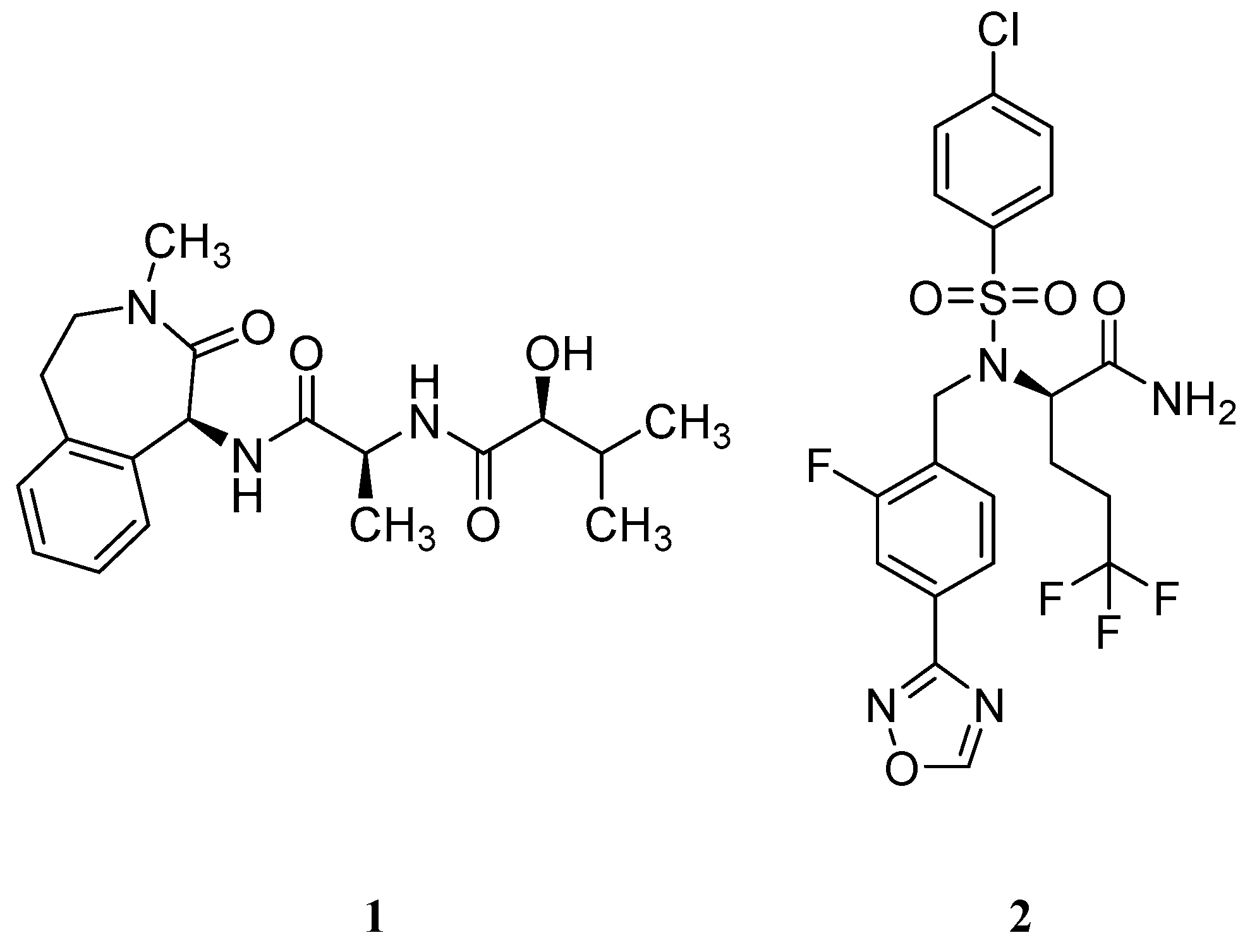
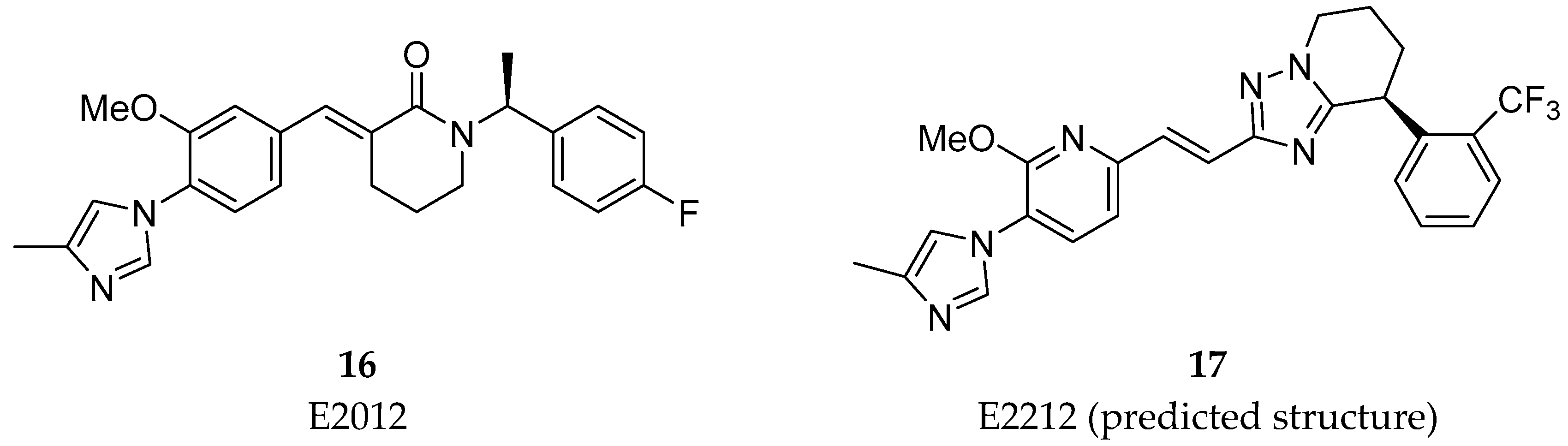
Non-NSAID Derived GSM’s
3.2. BACE-1 Inhibitors
3.2.1. BACE-1 Inhibitor Development
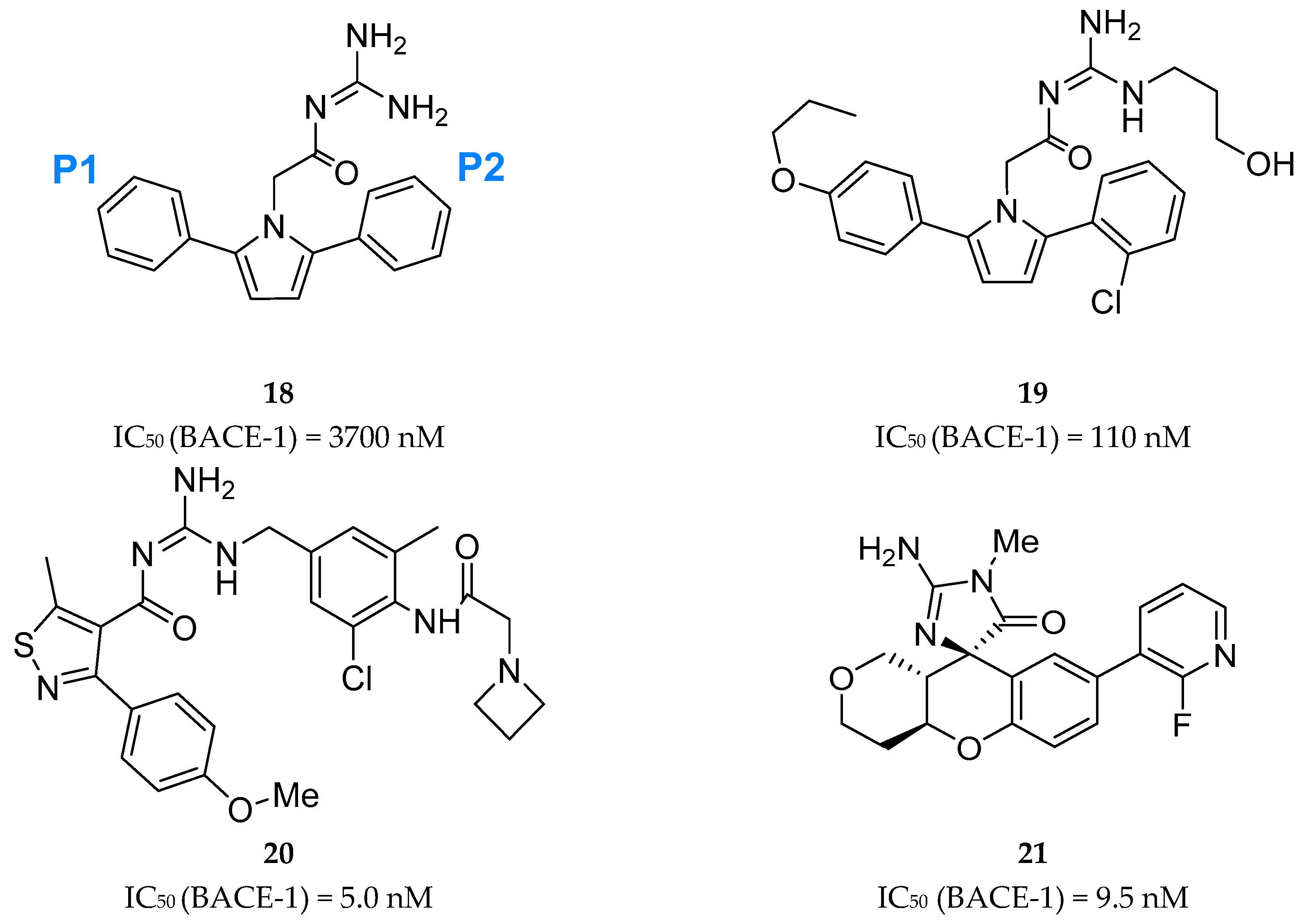
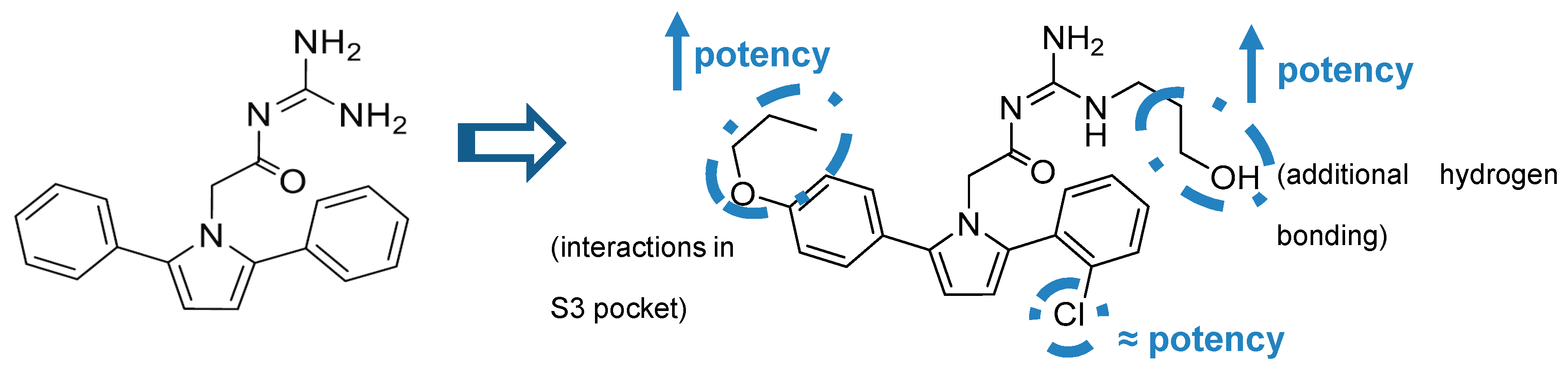
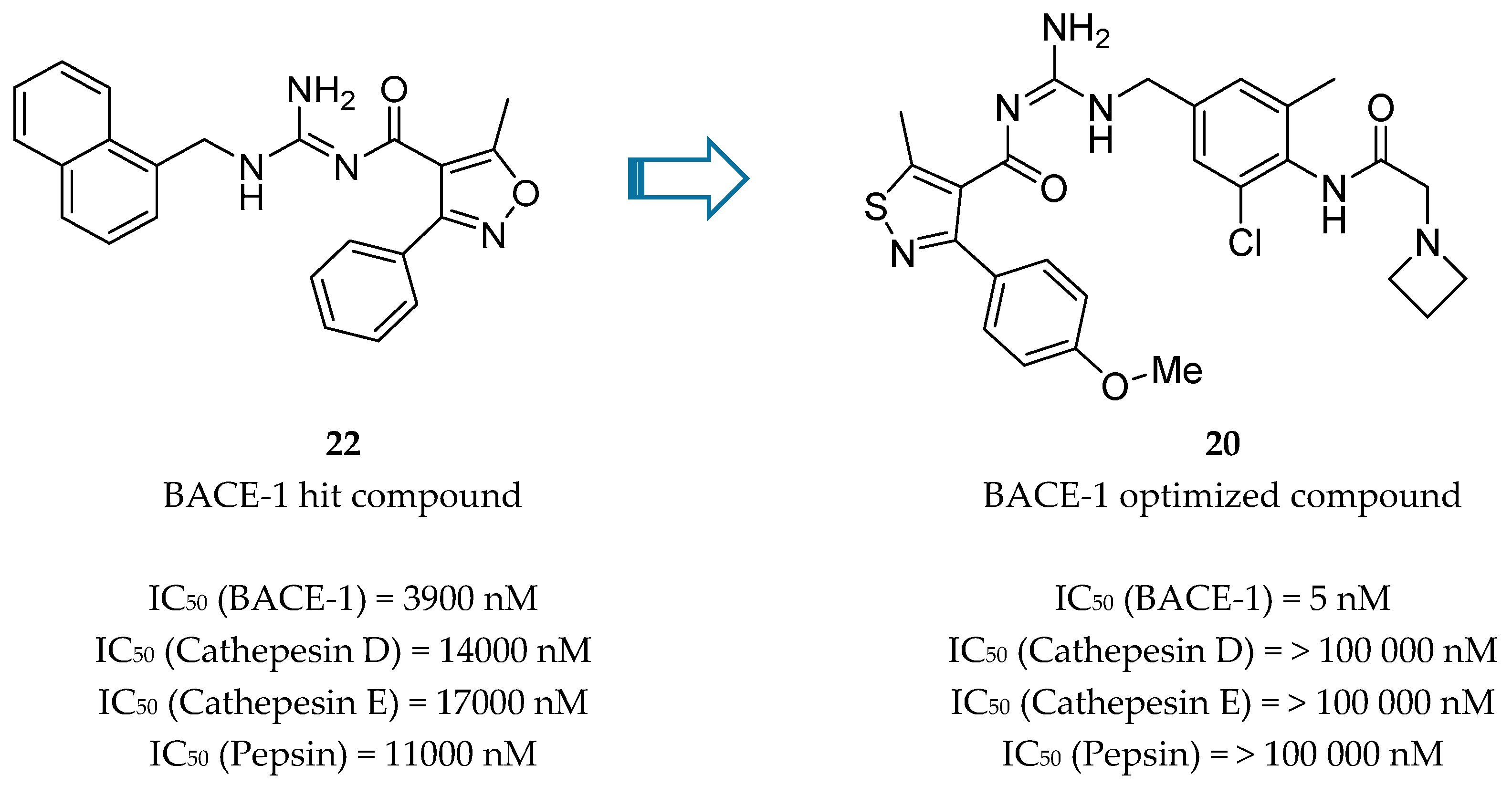
Acyl-Guanidine-Based Inhibitors
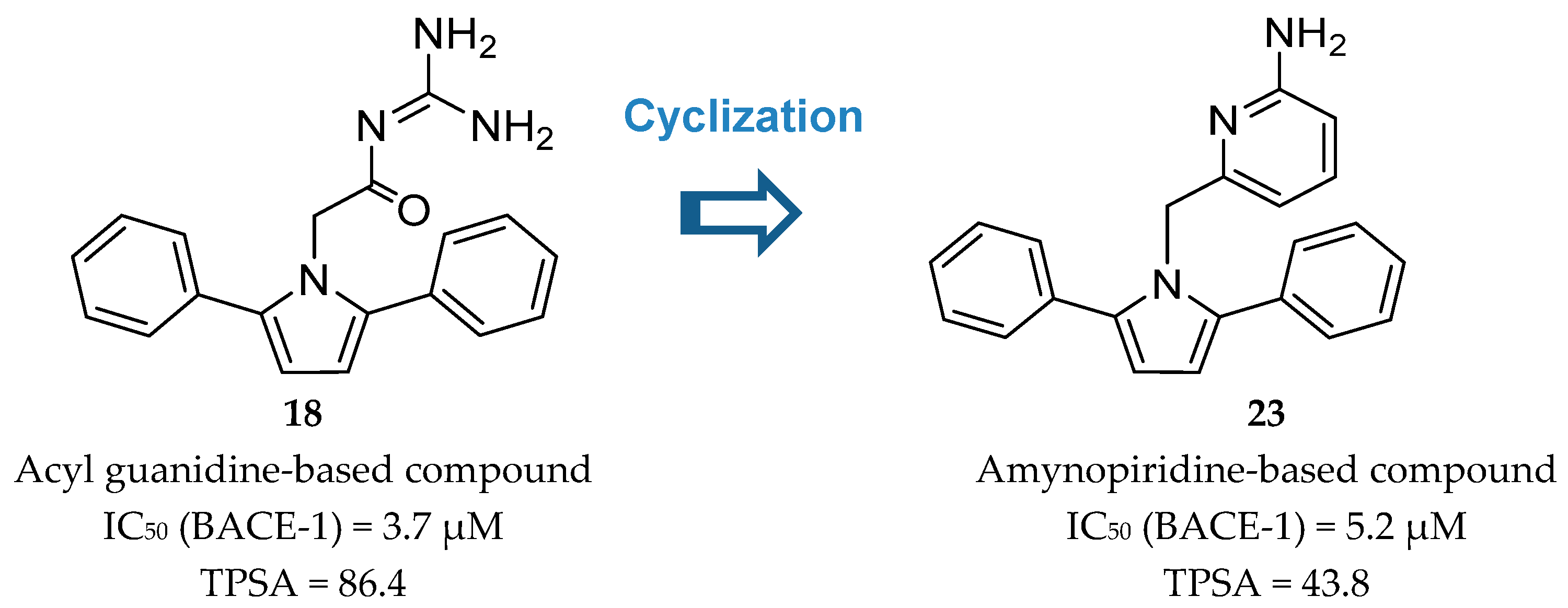
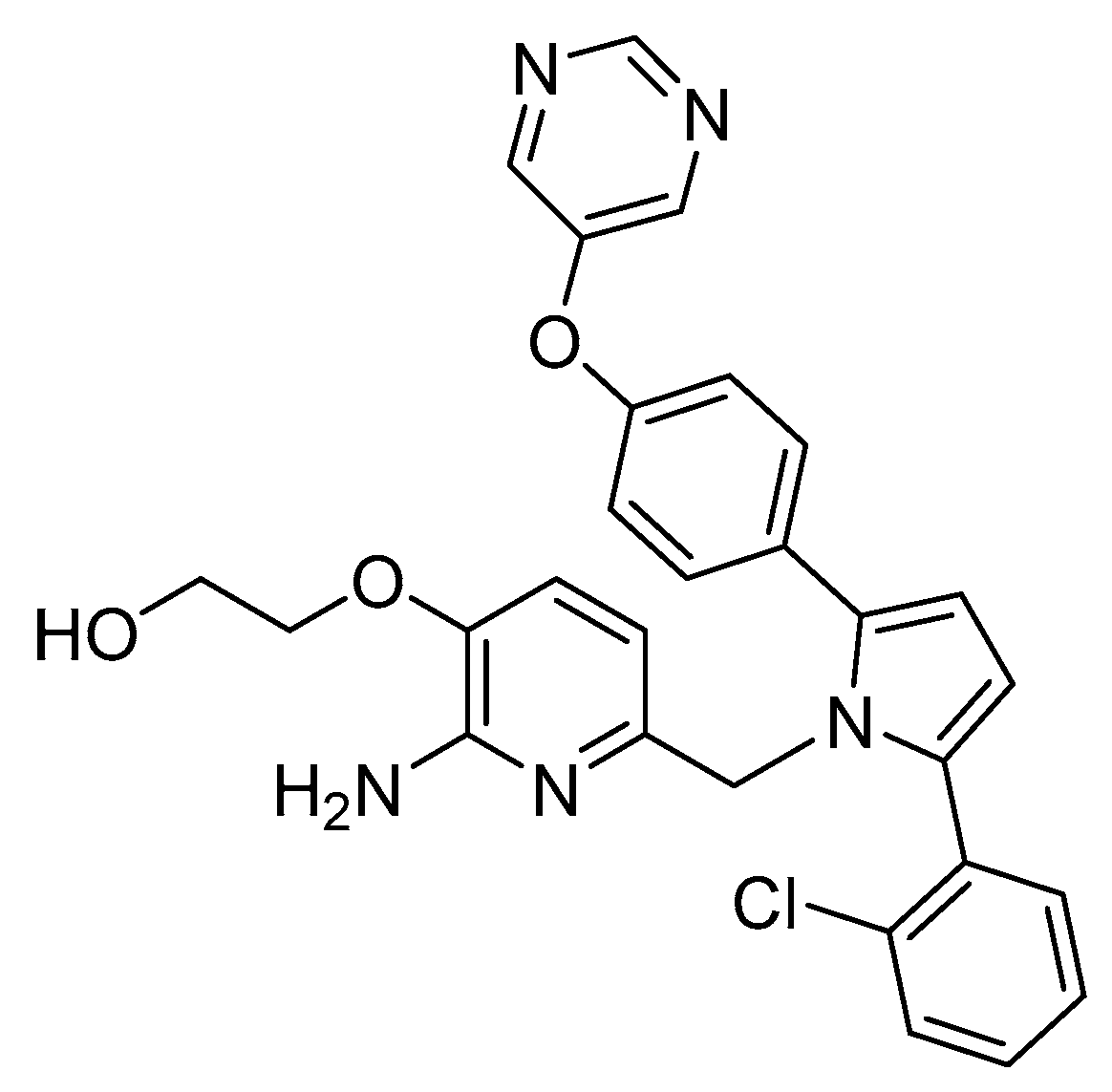
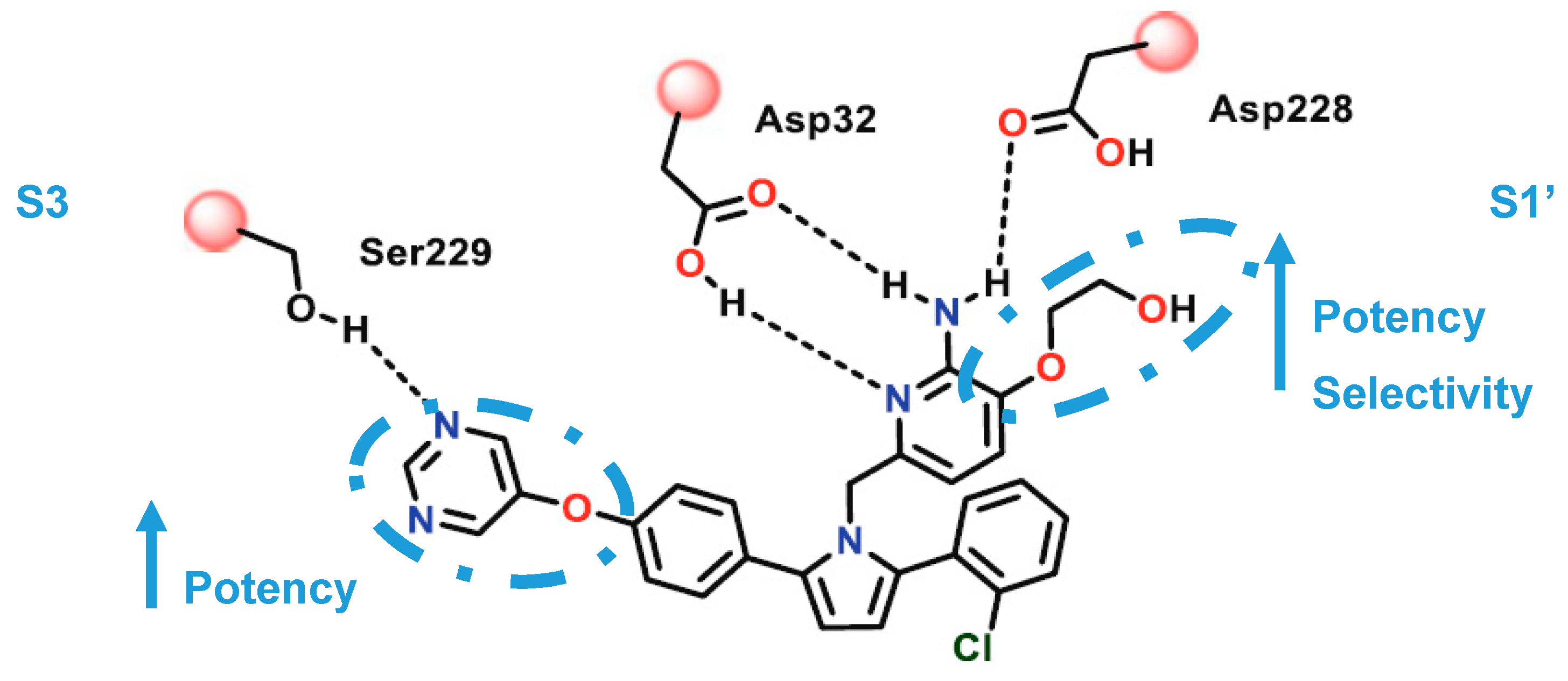
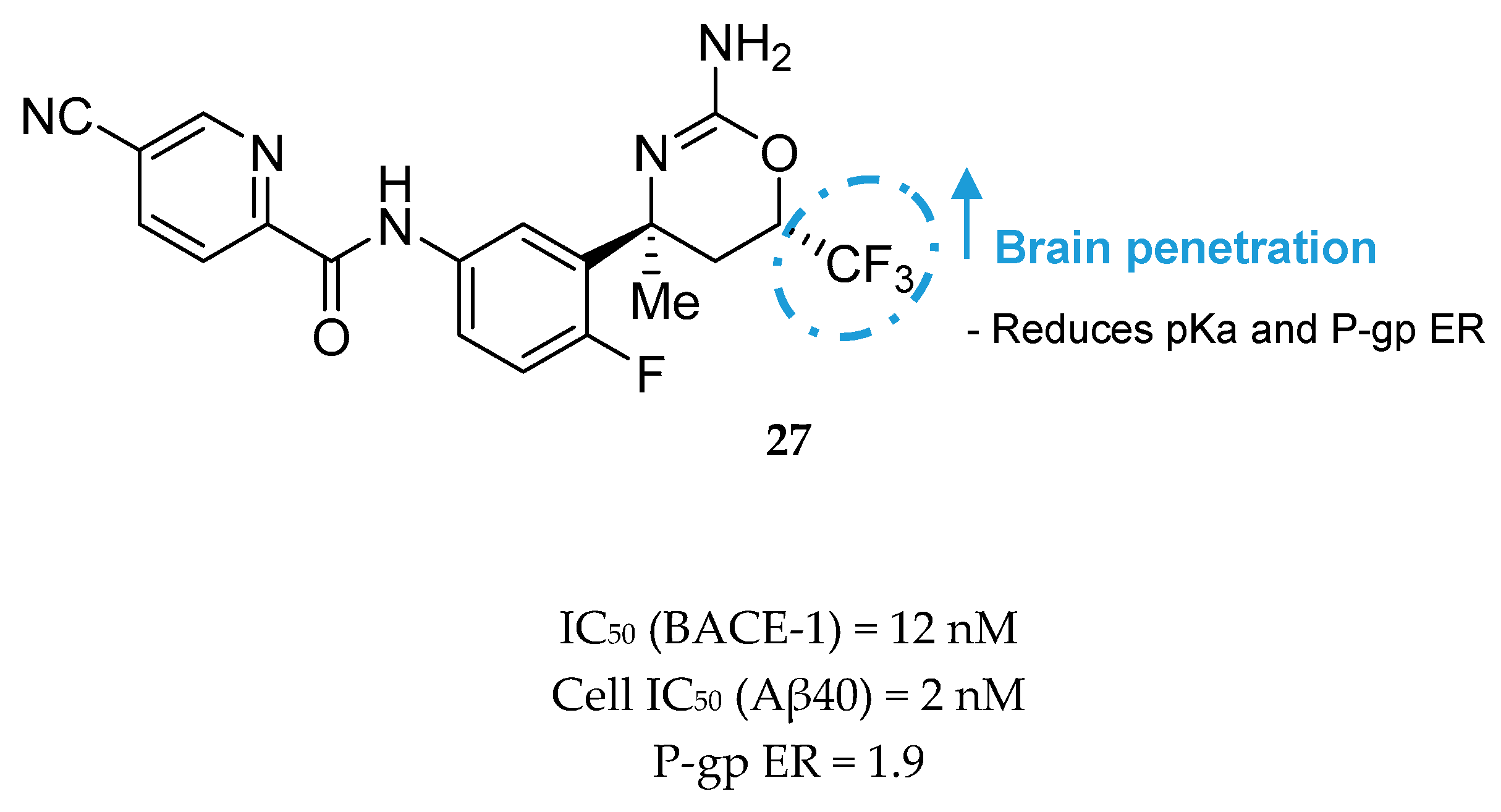
2-Aminopyridine-Based Inhibitors
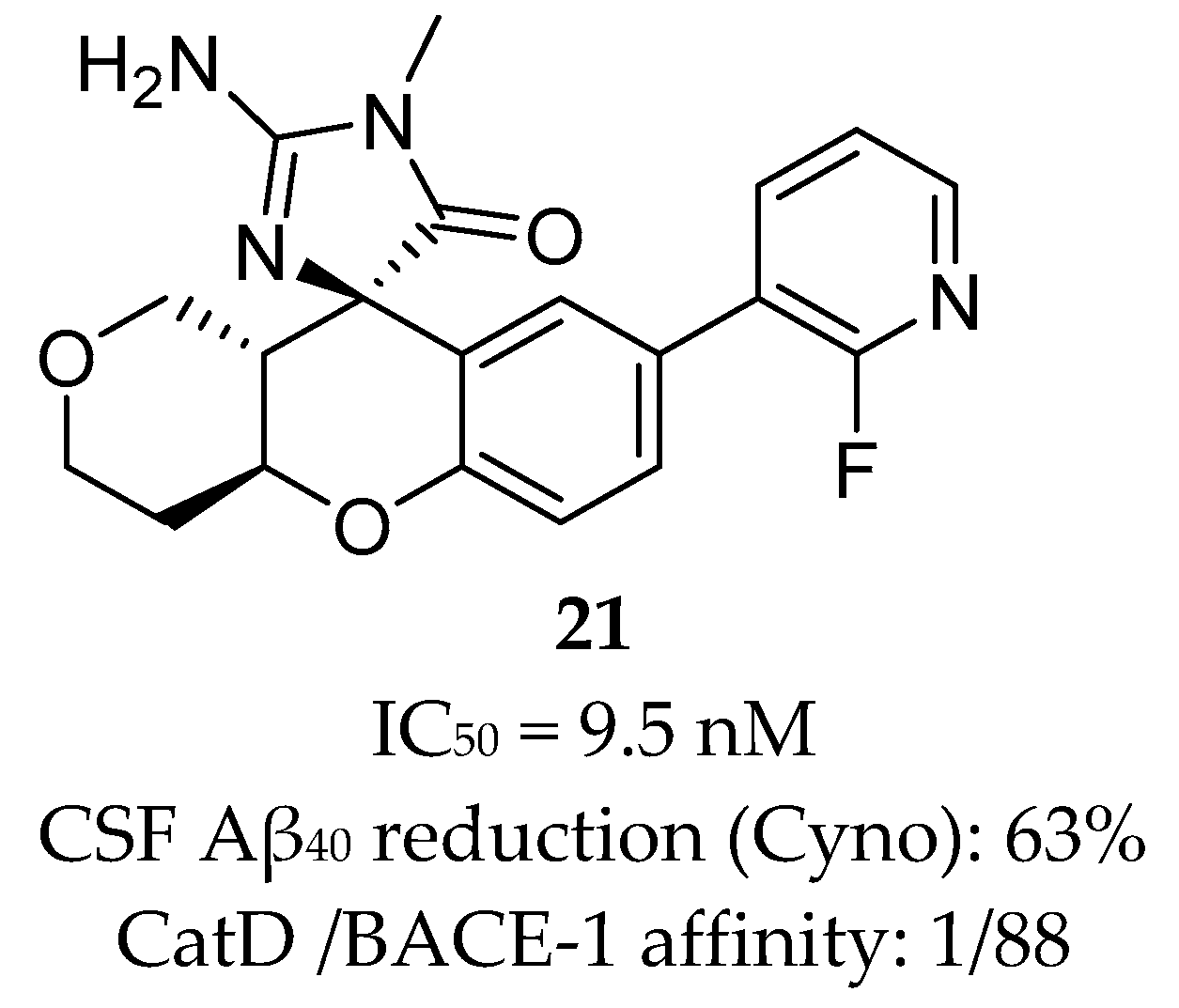
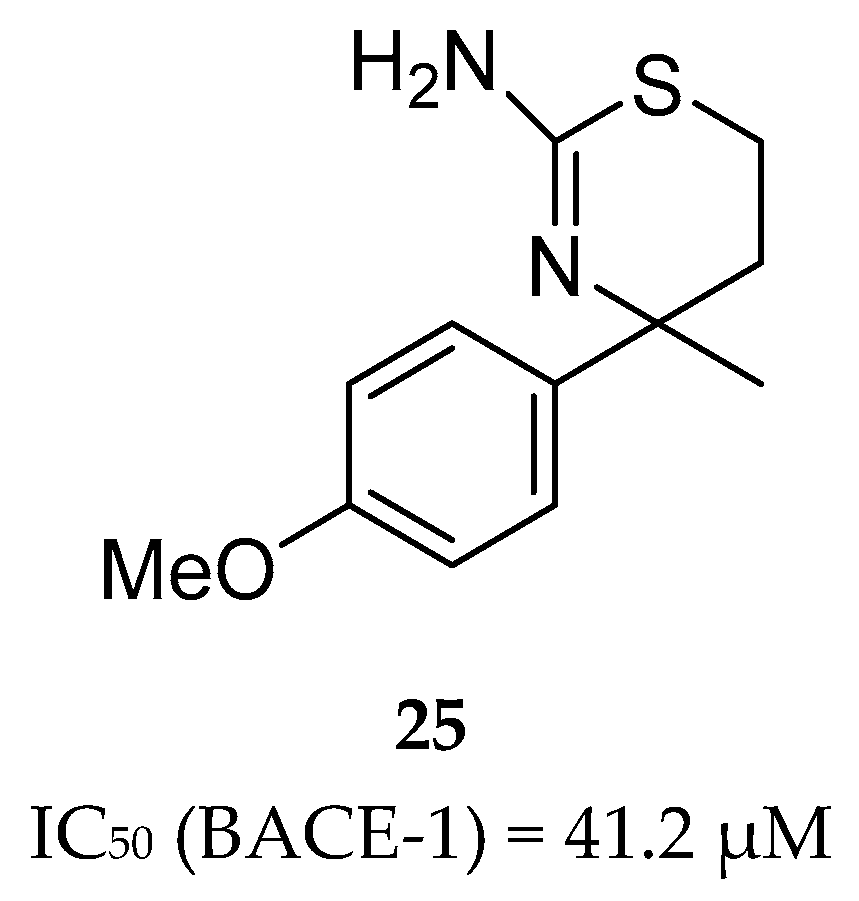
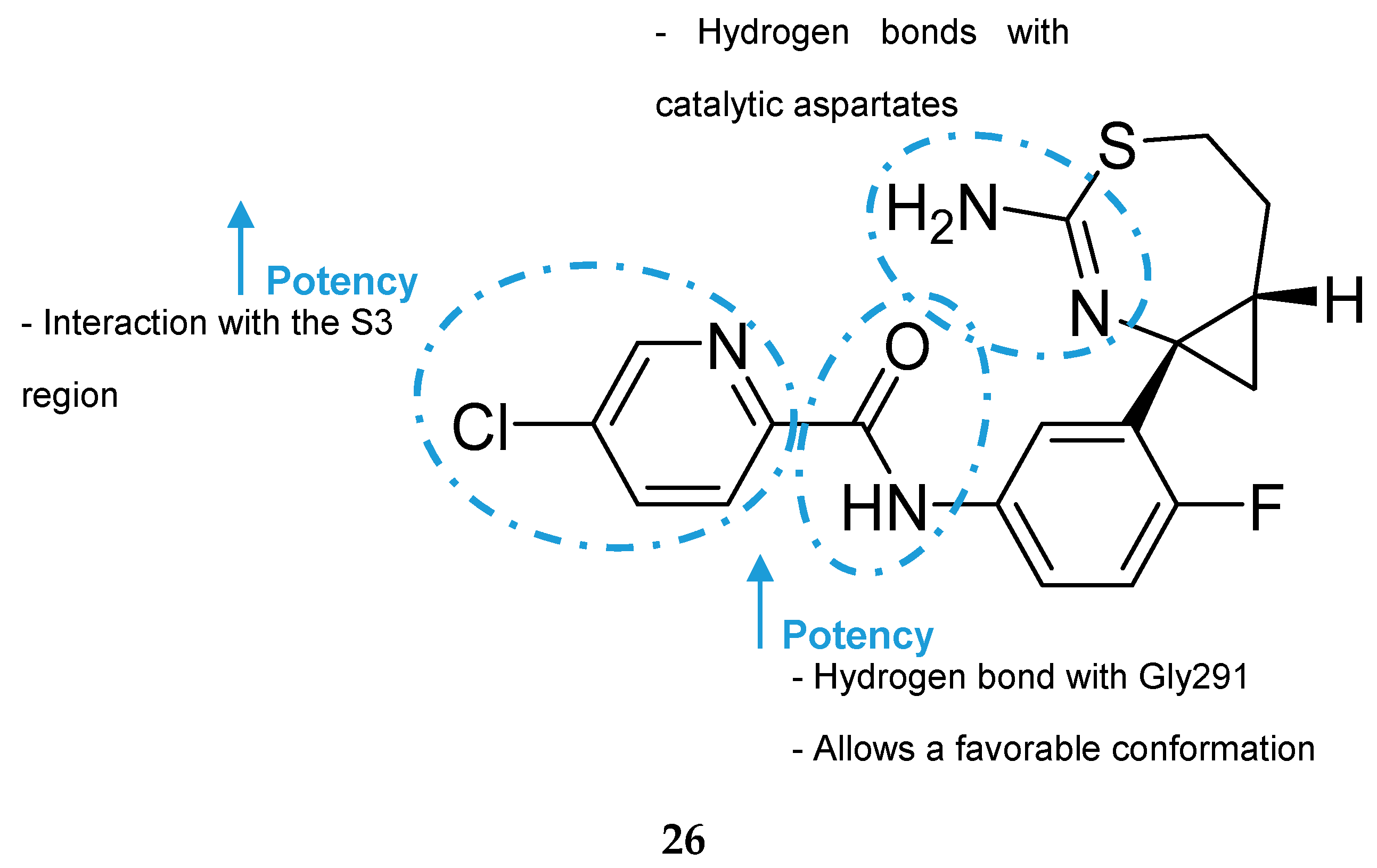
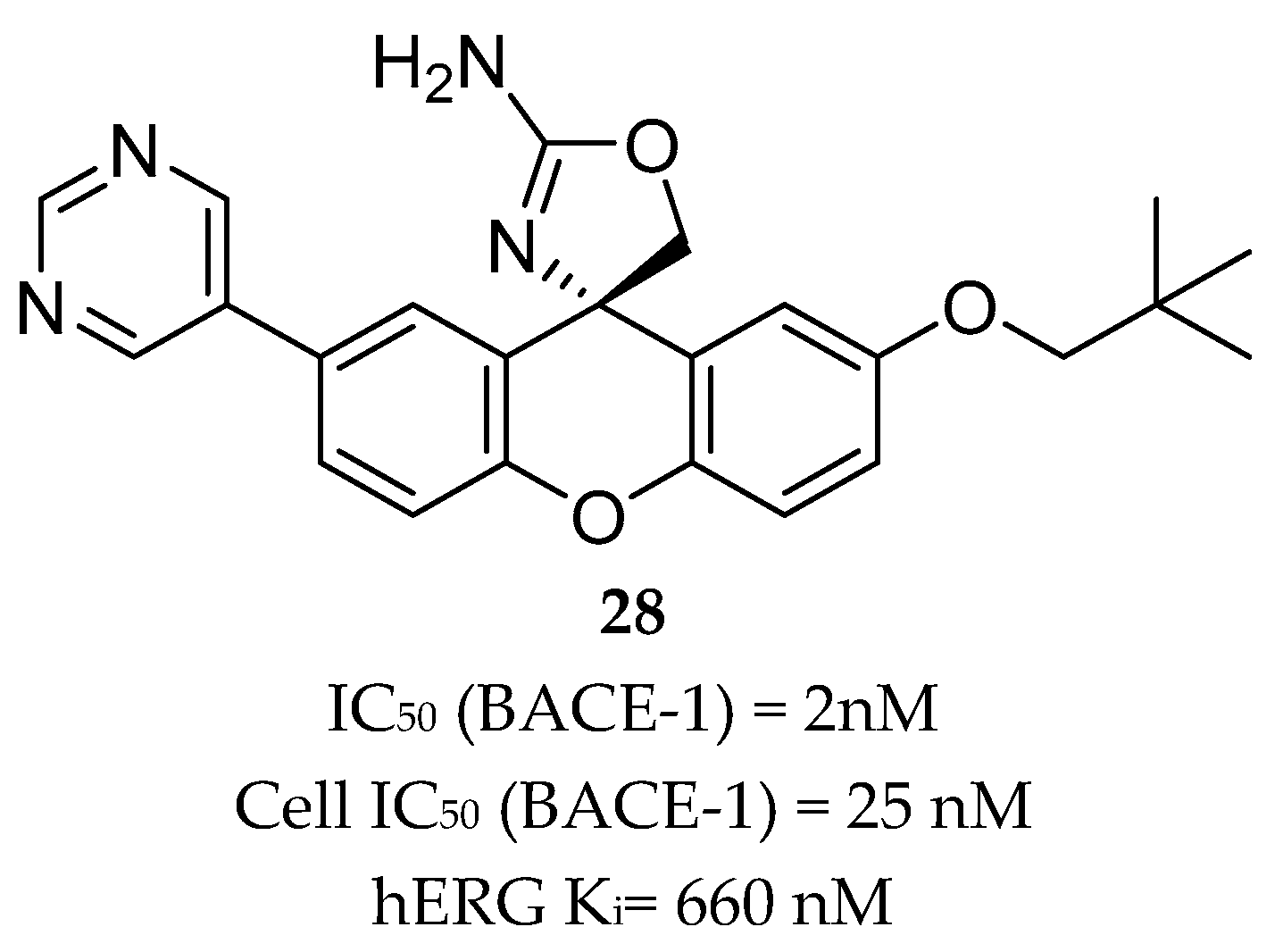
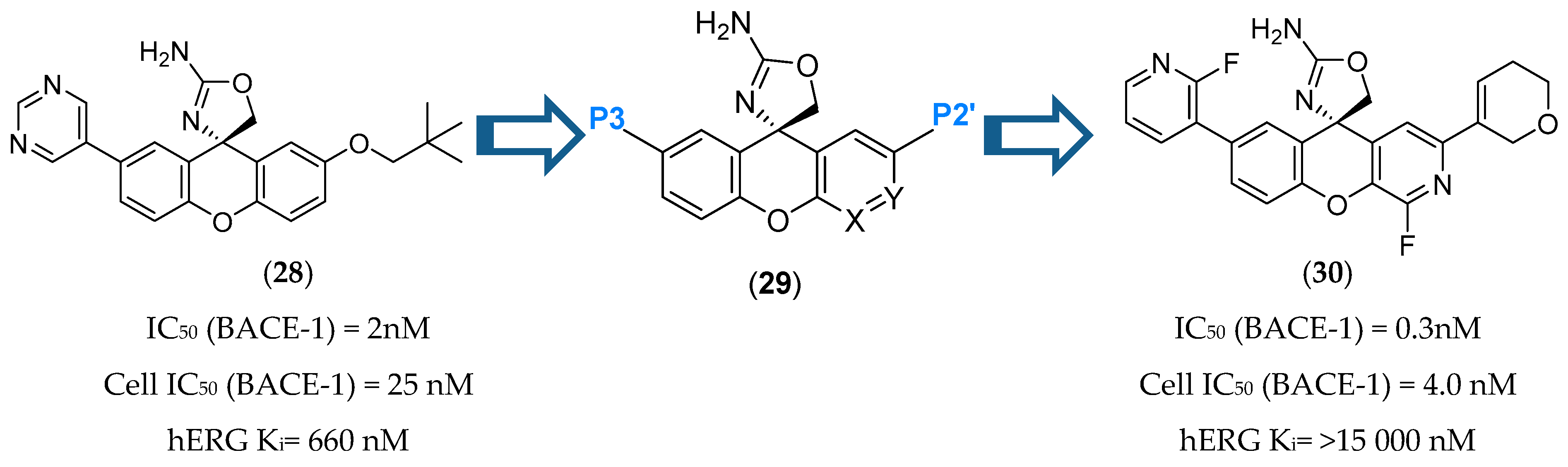
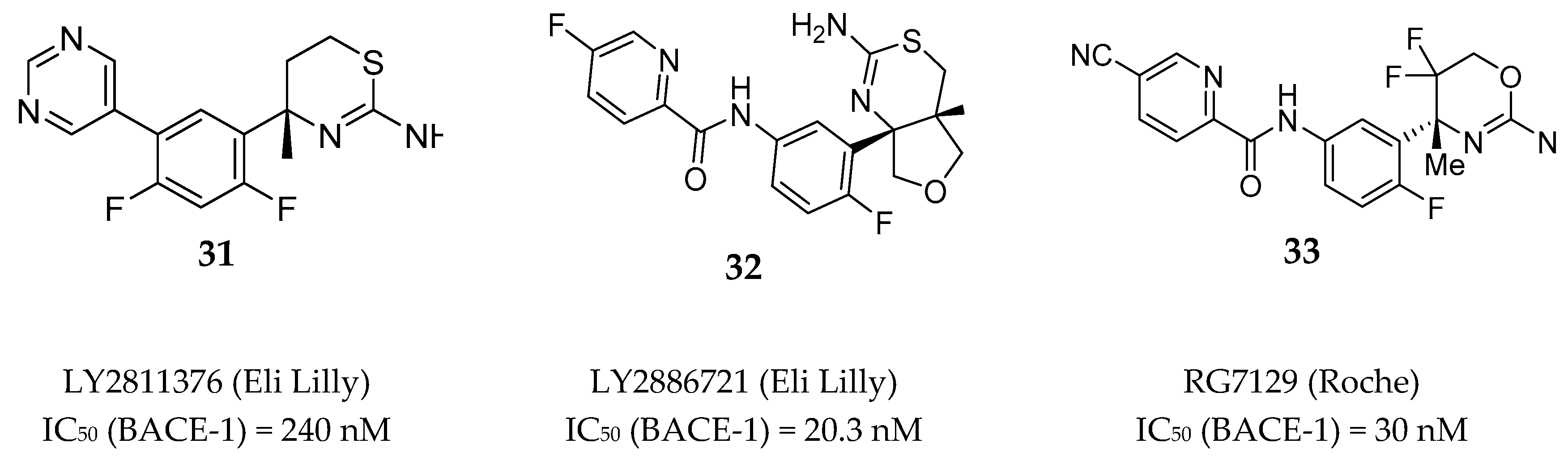
Aminothiazine- and Aminooxazoline-Based Inhibitors
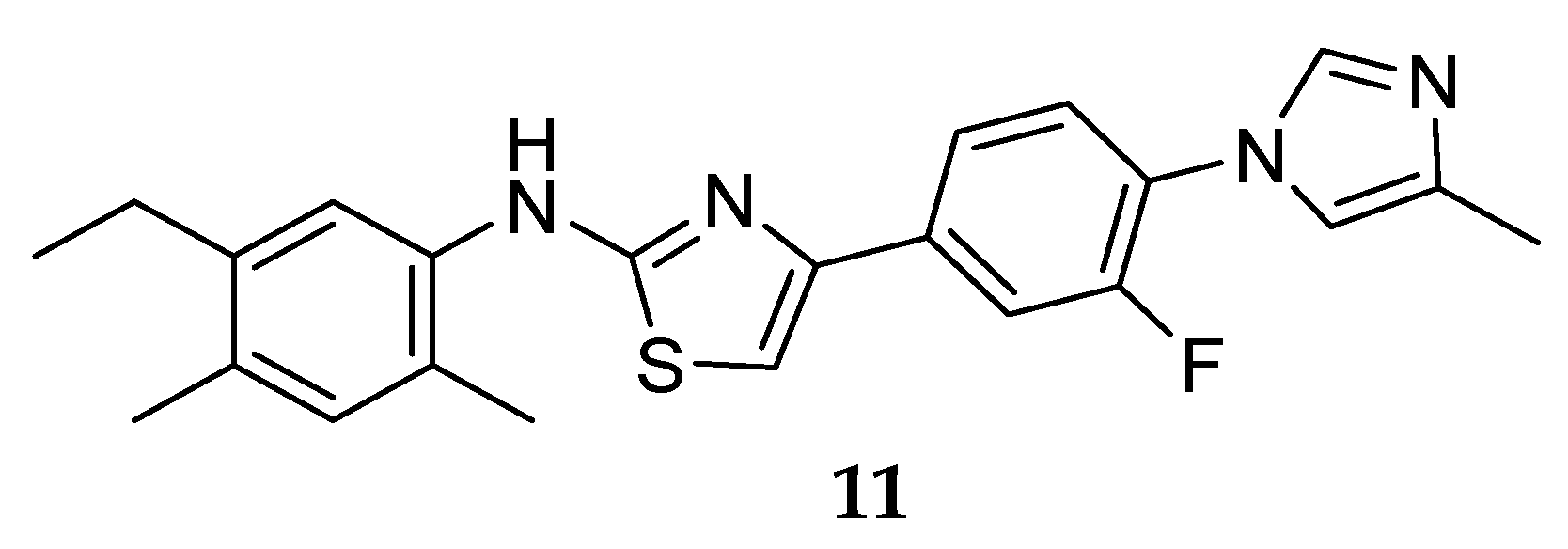
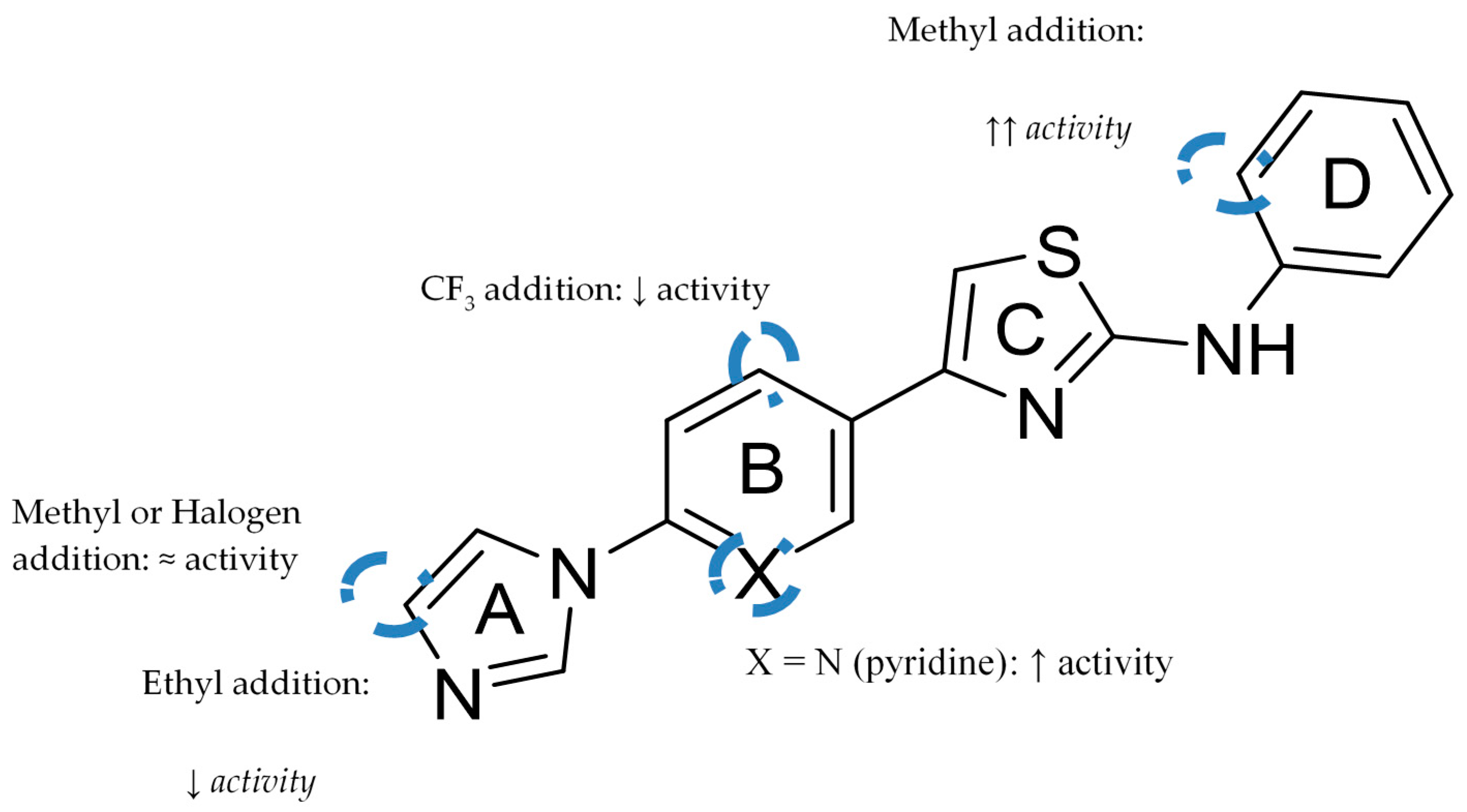
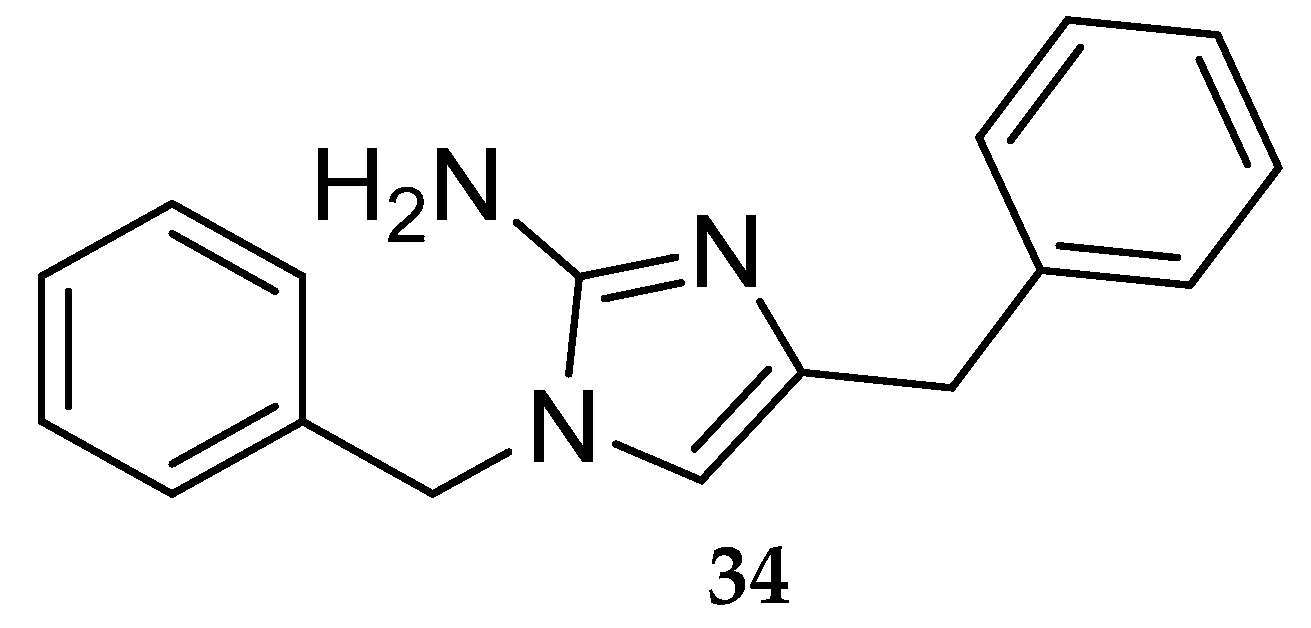
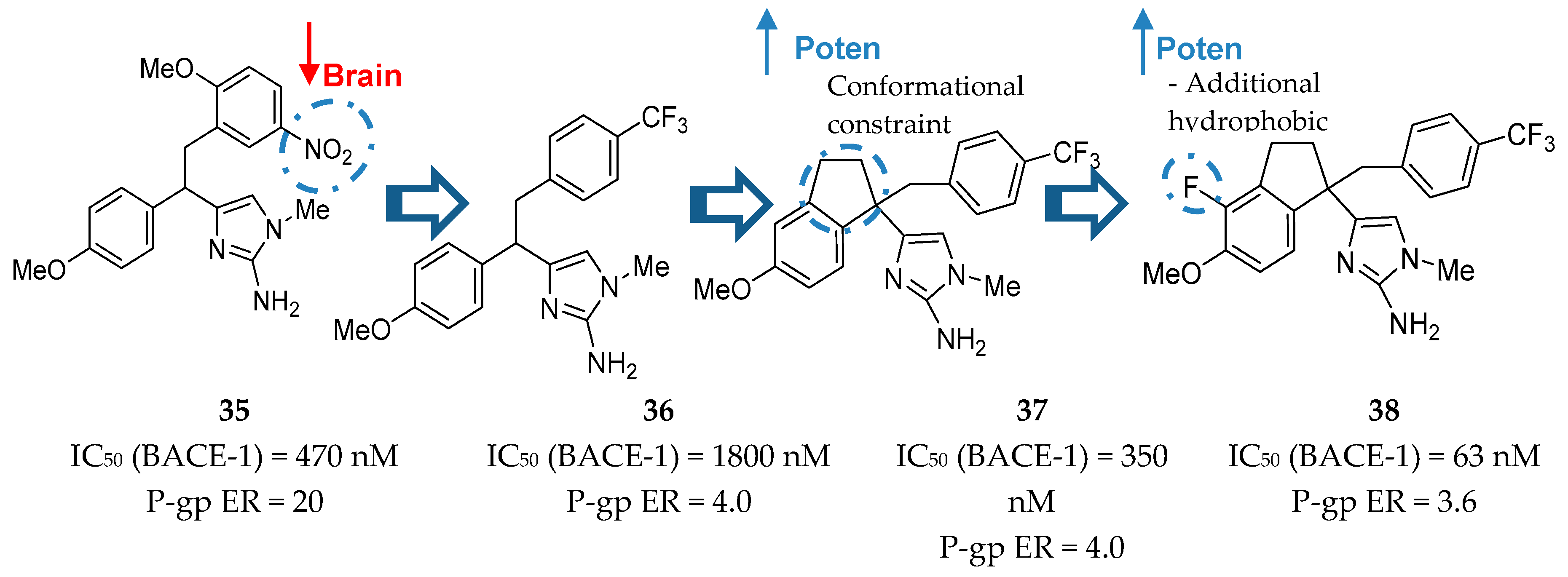
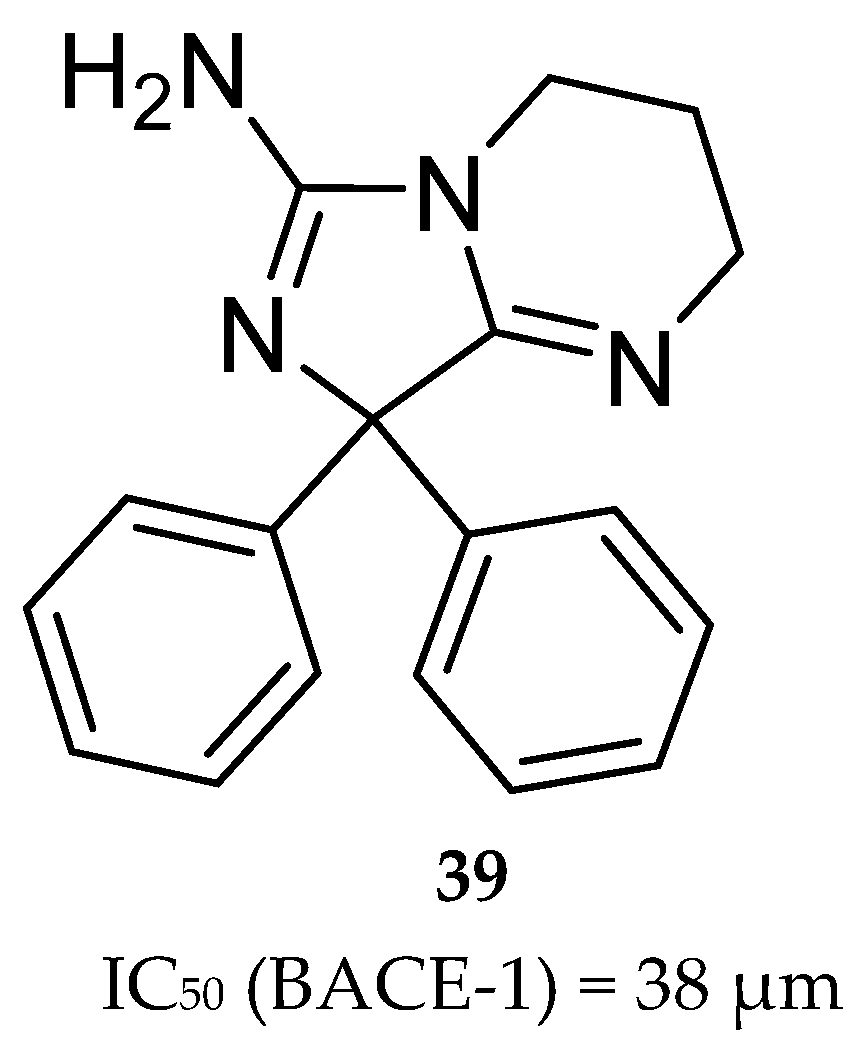
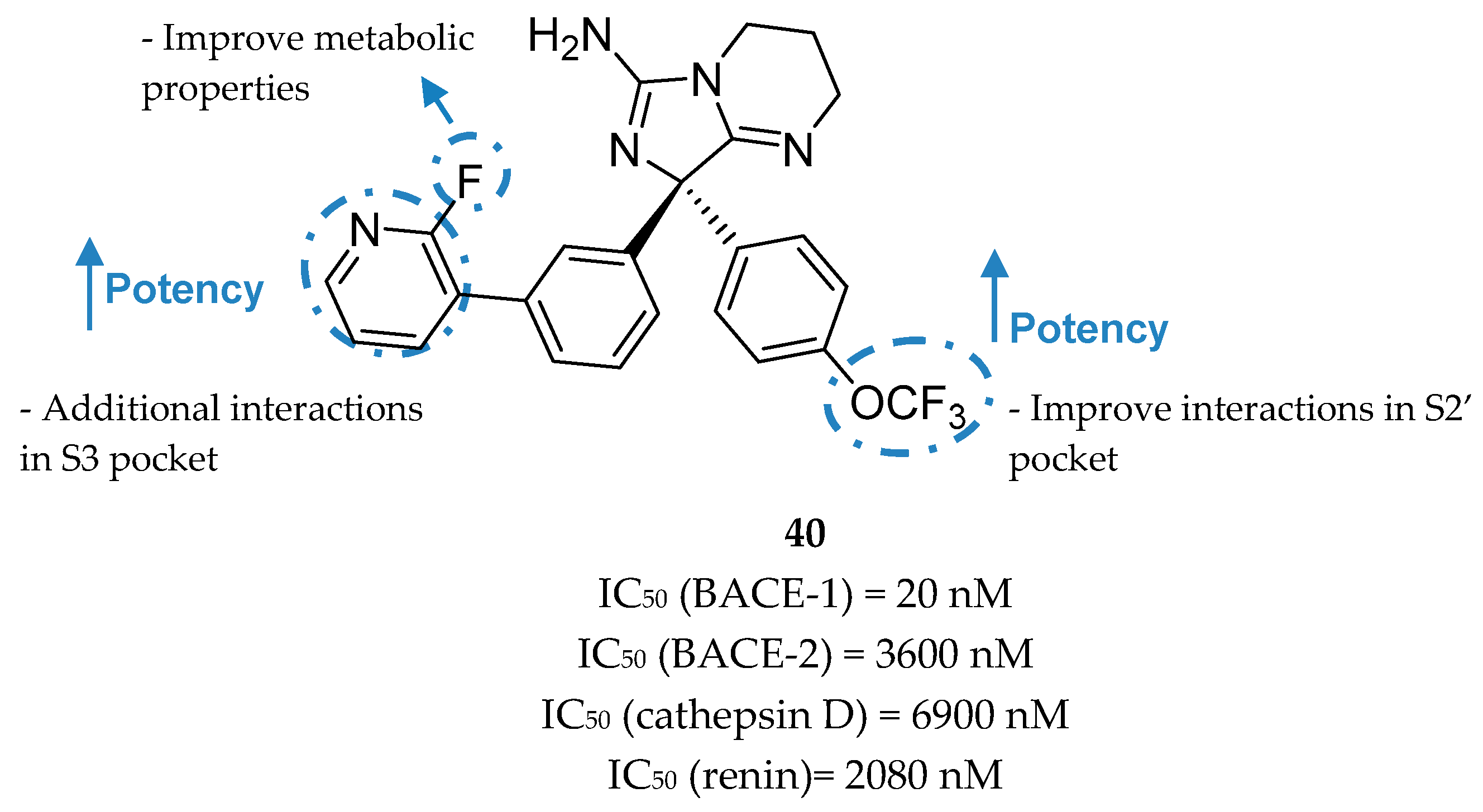
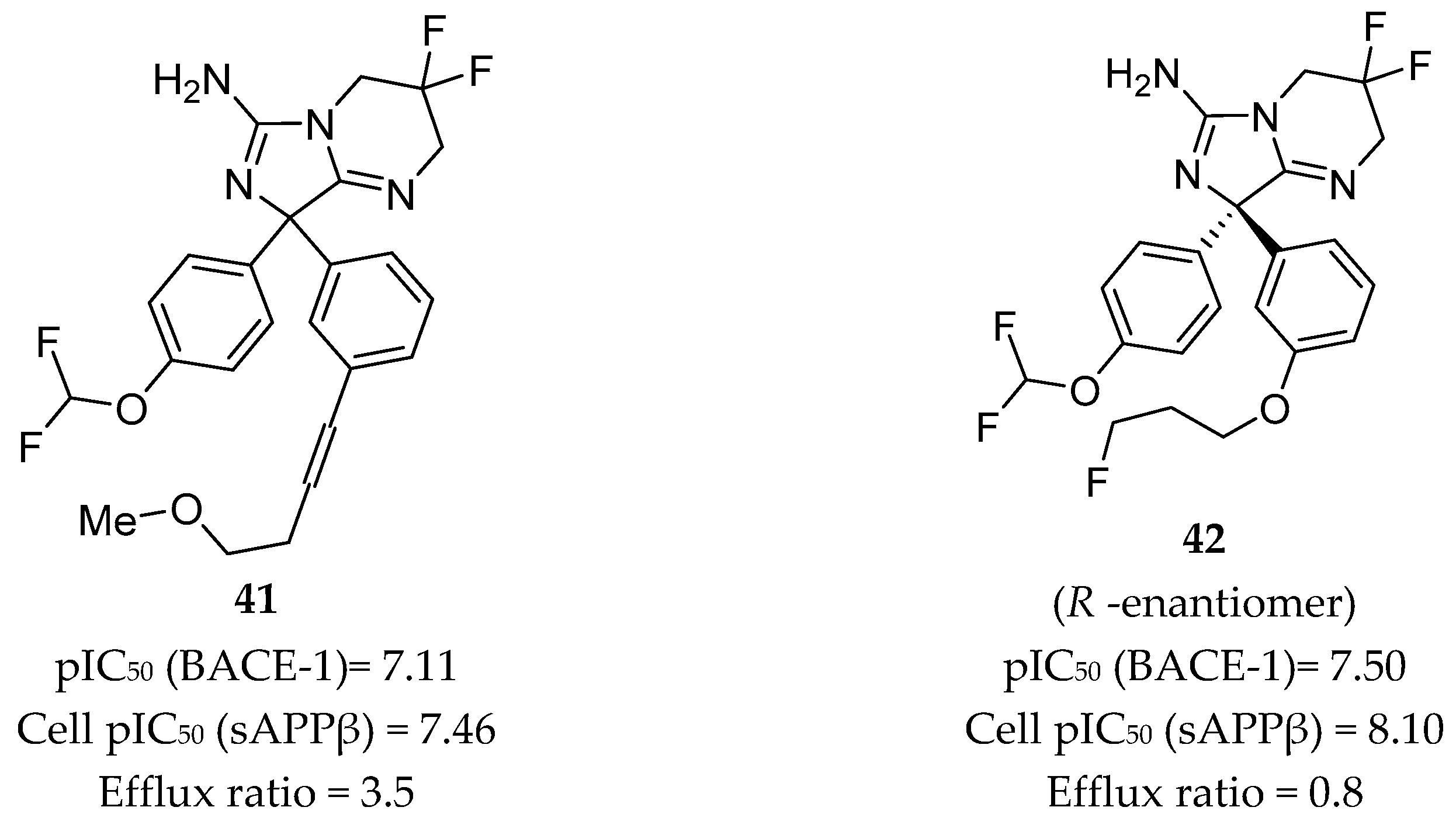
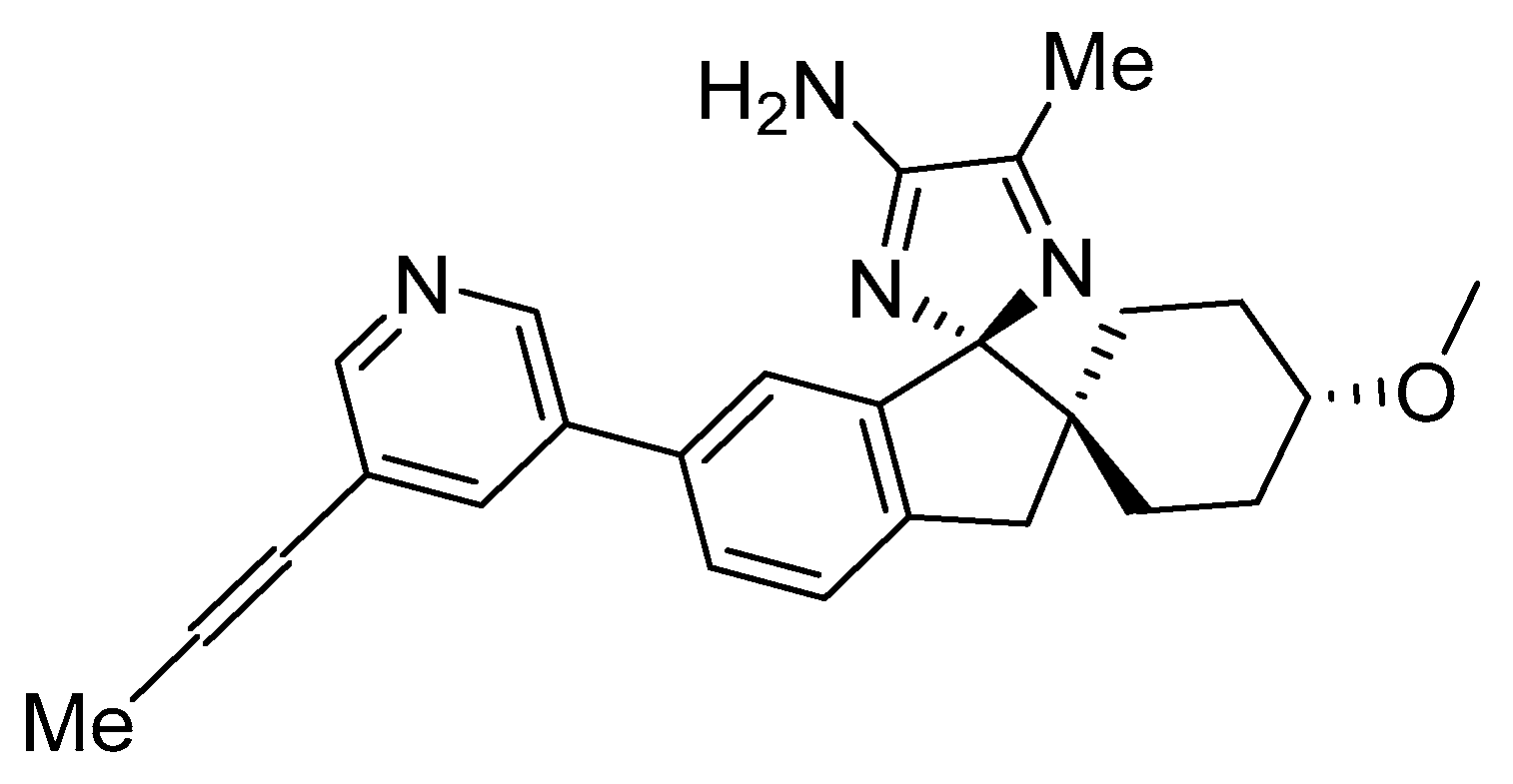
Aminoimidazole-Based Inhibitors
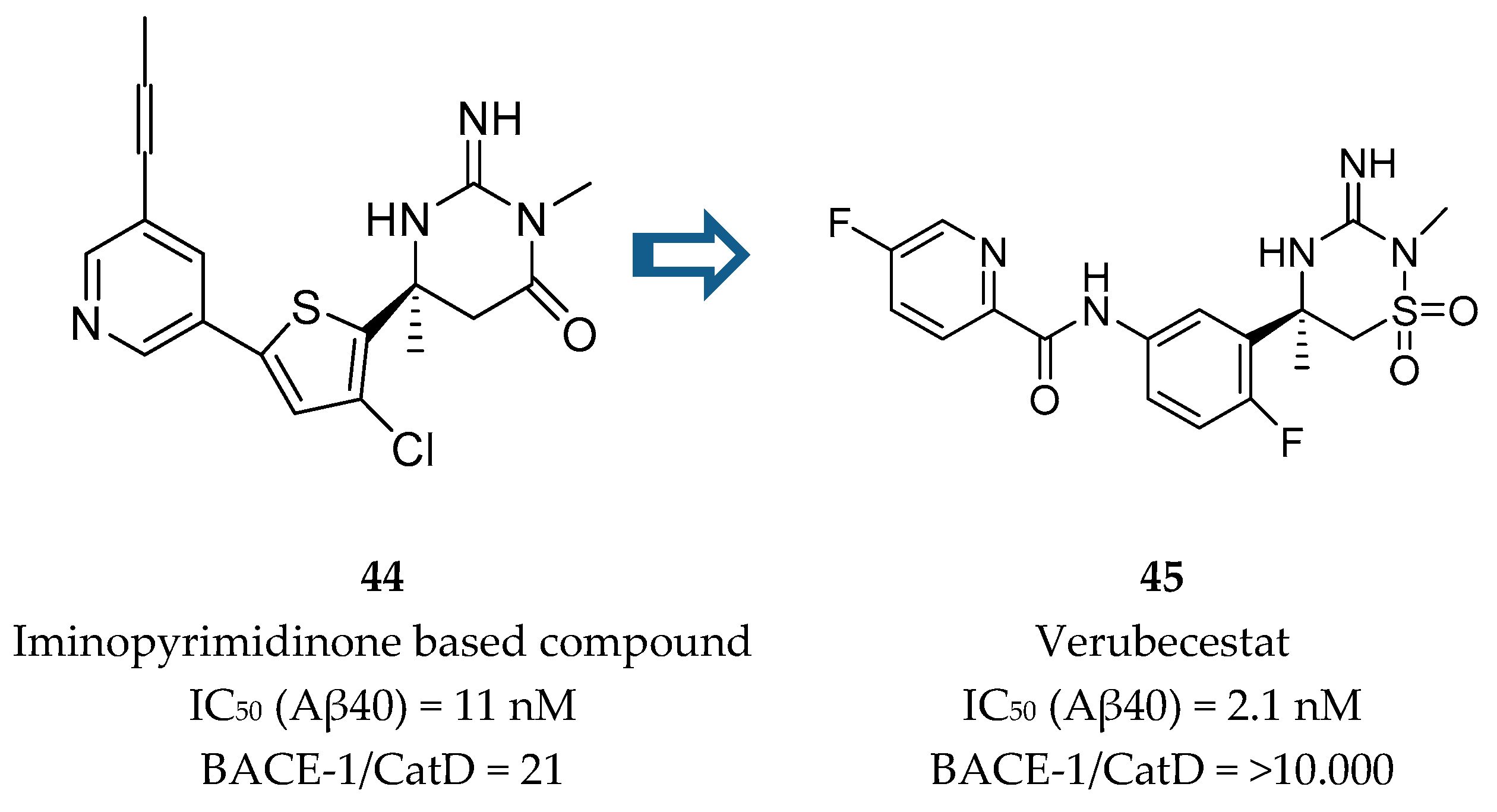
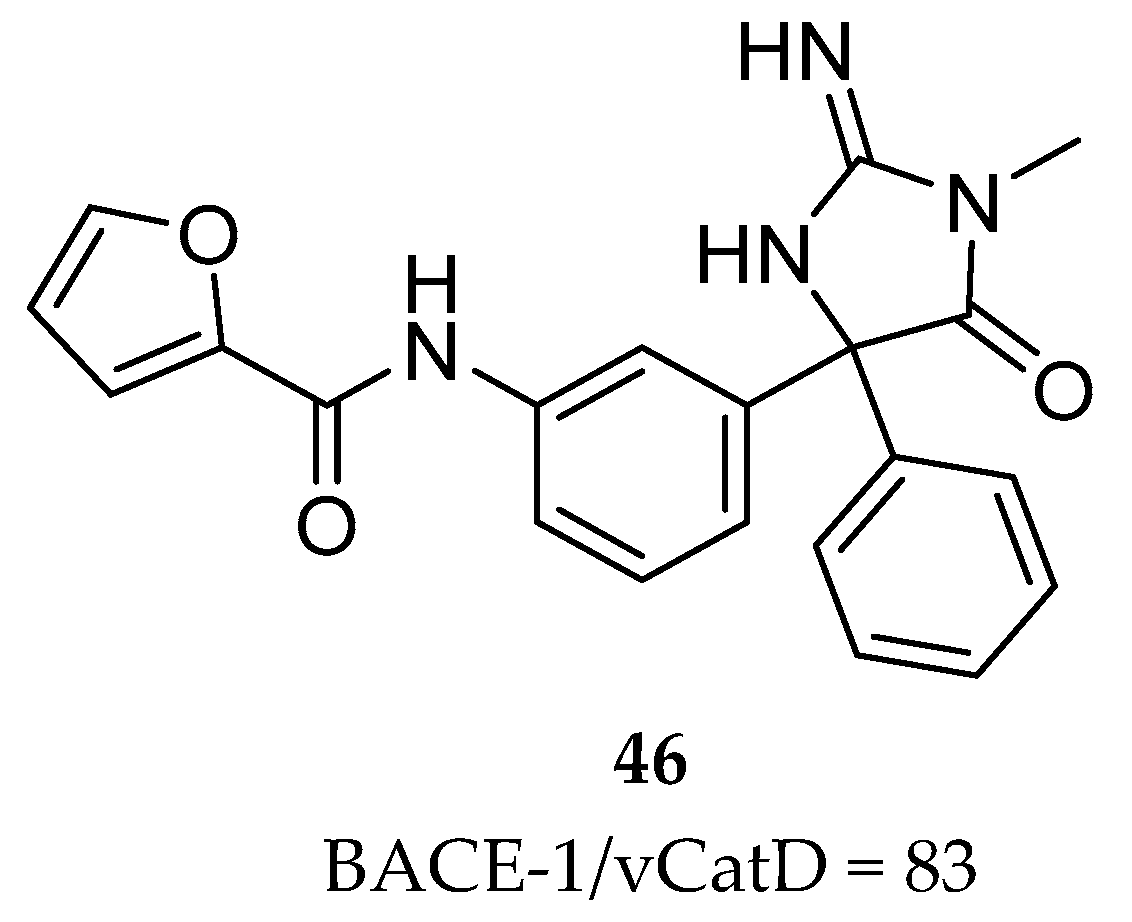
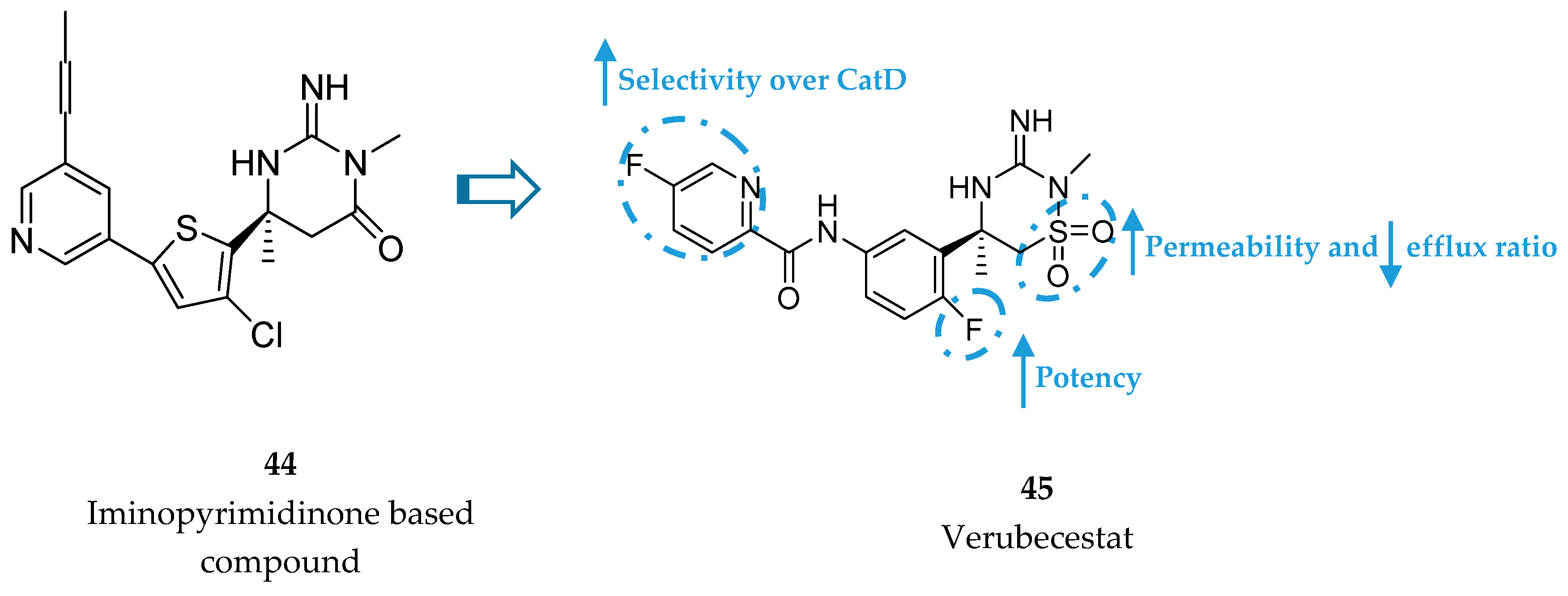
Iminothiadiazinane Dioxide-Based Inhibitors
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Duthey, B. Background Paper 6.11 Alzheimer Disease and Other Dementias, Update on 2004; World Health Organization: Geneva, Switzerland, February 2013; pp. 1–77. [Google Scholar]
- Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An english translation of alzheimer’s 1907 paper, “Uber eine eigenartige erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Eurostat. Population Structure and Ageing. Available online: http://ec.europa.eu/eurostat/statistics-explained/index.php/Population_structure_and_ageing (accessed on 5 January 2019).
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar]
- Schneider, J.; Arvanitakis, Z.; Leurgans, S.; Bennett, D. The Neuropathology of Probable Alzheimer’s Disease and Mild Cognitive Impairment. Ann. Neurol. 2010, 66, 200–208. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 1–7. [Google Scholar] [CrossRef]
- Balson, R.; Gibson, P.R.; Ames, D.; Bhathal, P.S. Tacrine-Induced Hepatotoxicity. CNS drugs 1995, 4, 168–169. [Google Scholar] [CrossRef]
- Scarpini, E.; Schelterns, P.; Feldman, H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol 2003, 2, 539–547. [Google Scholar] [CrossRef]
- FDA US Food and Drug Administration. Drug Approvals and Databases. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-487_Namenda.cfm (accessed on 5 January 2019).
- Alzheimer-Association. Changing the Trajectory of Alzheimer’s Disease: How a Treatment by 2025 Saves Lives and Dollars. Alzheimer’s Dement. 2013, 9, 208–245. [Google Scholar]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer ’s disease drug development pipeline: 2018. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 195–214. [Google Scholar] [CrossRef]
- Suzuki, K.; Wata, A.I.; Watsubo, T.I. The past, present, and future of disease-modifying therapies for Alzheimer ’s disease. Proc. Jpn. Acad. Ser. B 2017, 93, 757–771. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Cárdenas, E.L.; Osswald, H.L. The Design, Development, and Evaluation of BACE1 Inhibitors for the Treatment of Alzheimer’s Disease. Springer Int. Publ. Ag 2017, 24, 27–86. [Google Scholar]
- Bursavich, M.G.; Harrison, B.A.; Blain, J.F. Gamma secretase modulators: New Alzheimer’s drugs on the horizon? J. Med. Chem. 2016, 59, 7389–7409. [Google Scholar] [CrossRef]
- Brouwers, N.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of Alzheimer’s disease: An update. Ann. Med. 2008, 40, 562–583. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer‘s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Hamada, Y.; Kiso, Y. New directions for protease inhibitors directed drug discovery. Biopolymers 2016, 106, 563–579. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Sci. Compass 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Wolfe, M.S. γ-Secretase inhibitors and modulators for Alzheimer’s disease. J. Neurochem. 2012, 120, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woltering, T.J.; Wostl, W.; Hilpert, H.; Rogers-Evans, M.; Pinard, E.; Mayweg, A.; Göbel, M.; Banner, D.W.; Benz, J.; Travagli, M.; et al. BACE1 inhibitors: A head group scan on a series of amides. Bioorganic Med. Chem. Lett. 2013, 23, 4239–4243. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Diehl, T.S.; Narayanan, S.; Funamoto, S.; Ihara, Y.; De Strooper, B.; Steiner, H.; Haass, C.; Wolfe, M.S. Active γ-secretase complexes contain only one of each component. J. Biol. Chem. 2007, 282, 33985–33993. [Google Scholar] [CrossRef]
- Weidemann, A.; Eggert, S.; Reinhard, F.B.M.; Vogel, M.; Paliga, K.; Baier, G.; Masters, C.L.; Beyreuther, K.; Evin, G. A novel ε-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with notch processing. Biochemistry 2002, 41, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2815–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, M.S. Processive proteolysis by γ-secretase and the mechanism of Alzheimer’s disease. Biol. Chem. 2012, 393, 899–905. [Google Scholar] [CrossRef]
- Zhao, G.; Cui, M.Z.; Mao, G.; Dong, Y.; Tan, J.; Sun, L.; Xu, X. Gamma-Cleavage Is Dependent on zeta-Cleavage During the Proteolytic Processing of Amyloid Precursor Protein Within Its Transmembrane Domain. J. Biol. Chem. 2005, 280, 37689–37697. [Google Scholar] [CrossRef]
- Oehlrich, D.; Berthelot, D.J.C.; Gijsen, H.J.M. γ-Secretase modulators as potential disease modifying anti-Alzheimer’s drugs. J. Med. Chem. 2011, 54, 669–698. [Google Scholar] [CrossRef]
- Shimojo, M.; Sahara, N.; Mizoroki, T.; Funamoto, S.; Morishima-Kawashima, M.; Kudo, T.; Takeda, M.; Ihara, Y.; Ichinose, H.; Takashima, A. Enzymatic characteristics of I213T mutant presenilin-1/γ-secretase in cell models and knock-in mouse brains: Familial Alzheimer disease-linked mutation impairs γ-site cleavage of amyloid precursor protein C-terminal fragment beta. J. Biol. Chem. 2008, 283, 16488–16496. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Tagami, S.; Yanagida, K.; Takami, M.; Kodama, T.S.; Mori, K.; Nakayama, T.; Ihara, Y.; Takeda, M. γ-Secretase Modulators and Presenilin 1 Mutants Act Differently on Presenilin/γ-Secretase Function to Cleave Aβ42 and Aβ43. Cell Rep. 2013, 3, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. Embo J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szaruga, M.; Veugelen, S.; Benurwar, M.; Lismont, S.; Sepulveda-Falla, D.; Lleo, A.; Ryan, N.S.; Lashley, T.; Fox, N.C.; Murayama, S.; et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J. Exp. Med. 2015, 212, 2003–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretner, B.; Fukumori, A.; Gutsmiedl, A.; Page, R.M.; Luebbers, T.; Galley, G.; Baumann, K.; Haass, C.; Steiner, H. Attenuated Aβ42 responses to low potency γ-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J. Biol. Chem. 2011, 286, 15240–15251. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; Van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Woo, H.N.; Park, J.S.; Gwon, A.R.; Arumugam, T.V.; Jo, D.G. Alzheimer’s disease and Notch signaling. Biochem. Biophys. Res. Commun. 2009, 390, 1093–1097. [Google Scholar] [CrossRef]
- Gillman, K.W.; Starrett, J.E.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.E.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor. Acs Med. Chem. Lett. 2010, 1, 120–124. [Google Scholar] [CrossRef]
- Crump, C.J.; Castro, S.V.; Wang, F.; Pozdnyakov, N.; Ballard, T.E.; Sisodia, S.S.; Bales, K.R.; Johnson, D.S.; Li, Y.M. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 2012, 51, 7209–7211. [Google Scholar] [CrossRef]
- Reynaud, E. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nat. Educ. 2010, 3, 212–216. [Google Scholar]
- Eriksen, J.L.; Sagi, S.A.; Smith, T.E.; Weggen, S.; Das, P.; McLendon, D.C.; Ozols, V.V.; Jessing, K.W.; Zavitz, K.H.; Koo, E.H.; et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Aβ42 in vivo. J. Clin. Investig. 2003, 112, 440–449. [Google Scholar] [CrossRef]
- Imbimbo, B.P. Why did tarenflurbil fail in Alzheimer’s Disease? J. Alzheimer’s Dis. 2009, 17, 757–760. [Google Scholar] [CrossRef]
- Peretto, I.; Radaelli, S.; Parini, C.; Zandi, M.; Raveglia, L.F.; Dondio, G.; Fontanella, L.; Misiano, P.; Bigogno, C.; Rizzi, A.; et al. Synthesis and biological activity of flurbiprofen analogues as selective inhibitors β-Amyloid1-42 secretion. J. Med. Chem. 2005, 48, 5705–5720. [Google Scholar] [CrossRef]
- Baumann, S.; Höttecke, N.; Schubenel, R.; Baumann, K.; Schmidt, B. NSAID-derived γ-secretase modulators. Part III: Membrane anchoring. Bioorganic Med. Chem. Lett. 2009, 19, 6986–6990. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Frigerio, E.; Breda, M.; Fiorentini, F.; Fernandez, M.; Sivilia, S.; Giardino, L.; Calzà, L.; Norris, D.; Casula, D.; et al. Pharmacokinetics and pharmacodynamics of CHF5074 after short-term administration in healthy subjects. Alzheimer Dis Assoc Disord 2013, 27, 278–286. [Google Scholar] [CrossRef]
- Van Vleet, T.R.; Liu, H.; Lee, A.; Blomme, E.A.G. Acyl glucuronide metabolites: Implications for drug safety assessment. Toxicol. Lett. 2017, 272, 1–7. [Google Scholar] [CrossRef]
- Kounnas, M.Z.; Lane-Donovan, C.; Nowakowski, D.W.; Herz, J.; Comer, W.T. NGP 555, a γ-secretase modulator, lowers the amyloid biomarker, Aβ42 in cerebrospinal fluid while preventing Alzheimer’s disease cognitive decline in rodents. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 65–73. [Google Scholar]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef]
- Kimura, T.; Kawano, K.; Doi, E.; Kitazawa, N.; Shin, K.; Miyagawa, T.; Kaneco, T.; Ito, K.; Takaishi, M.; Sasaki, T.; et al. Cinnamide Compound. Patent Application Publication No. WO 2005115990 A1, 8 December 2015. [Google Scholar]
- Hashimoto, T.; Ishibashi, A.; Hagiwara, H.; Murata, Y.; Takenaka, O.; Miyagawa, T. E2012: A novel gamma-secretase modulator-pharmacology part. Alzheimer’s Dement. 2010, 6, S242. [Google Scholar] [CrossRef]
- Boggs, L.N.; Lindstrom, T.; Watson, B.; Sheehan, S.; Audia, J.E.; May, P.C.; Lilly, E. E2012: A novel gamma-secretase modulator-mechanism of action. Alzheimer’s Dement. 2010, 6, S541–S542. [Google Scholar] [CrossRef]
- Nagy, C.; Schuck, E.; Ishibashi, A.; Nakatani, Y.; Rege, B.; Logovinsky, V. E2012, a novel gamma-secretase modulator, decreases plasma amyloid-beta (Aβ) levels in humans. Alzheimer’s Dement. 2010, 6, S574. [Google Scholar] [CrossRef]
- Nakano-Ito, K.; Fujikawa, Y.; Hihara, T.; Shinjo, H.; Kotani, S.; Suganuma, A.; Aoki, T.; Tsukidate, K. E2012-induced cataract and its predictive biomarkers. Toxicol. Sci. 2014, 137, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Logovinsky, V.; Schuck, E.; Kaplow, J.; Chang, M.K.; Miyagawa, T.; Wong, N.; Ferry, J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel γ-secretase modulator, E2212, in healthy human subjects. J. Clin. Pharmacol. 2014, 54, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Stepan, A.F.; Kauffman, G.W.; Johnson, D.S. Novel γ-secretase modulators for the treatment of Alzheimer’s disease: A review focusing on patents from 2010 to 2012. Expert Opin. Ther. Pat. 2013, 23, 1349–1366. [Google Scholar] [CrossRef] [PubMed]
- Voytyuk, I.; de Strooper, B.; Chávez-Gutiérrez, L. Modulation of γ- and β-Secretases as Early Prevention Against Alzheimer ’ s Disease. Biol. Psychiatry 2018, 83, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Rogers, G.; Citron, M. β-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Min, K.; Kwak, H.S.; Koo, K.D.; Lim, D.; Seo, S.W.; Choi, J.U.; Platt, B.; Choi, D.Y. Synthesis, SAR, and X-ray structure of human BACE-1 inhibitors with cyclic urea derivatives. Bioorganic Med. Chem. Lett. 2008, 18, 2900–2904. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.C.; Manas, E.S.; Stock, J.R.; Condon, J.S.; Jennings, L.D.; Aulabaugh, A.; Chopra, R.; Cowling, R.; Ellingboe, J.W.; Fan, K.Y.; et al. Acylguanidines as small-molecule β-secretase inhibitors. J. Med. Chem. 2006, 49, 6158–6161. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, M.; Correa-Basurto, J.; Gutiérrez, A.; Vitorica, J.; Rosales-Hernández, M.C. Asp32 and Asp228 determine the selective inhibition of BACE1 as shown by docking and molecular dynamics simulations. Eur. J. Med. Chem. 2016, 124, 1142–1154. [Google Scholar] [CrossRef]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar] [CrossRef]
- Gerritz, S.W.; Zhai, W.; Shi, S.; Zhu, S.; Toyn, J.H.; Meredith, J.E.; Iben, L.G.; Burton, C.R.; Albright, C.F.; Good, A.C.; et al. Acyl guanidine inhibitors of β-secretase (BACE-1): Optimization of a micromolar hit to a nanomolar lead via iterative solid- and solution-phase library synthesis. J. Med. Chem. 2012, 55, 9208–9223. [Google Scholar] [CrossRef]
- Thomas, A.A.; Hunt, K.W.; Volgraf, M.; Watts, R.J.; Liu, X.; Vigers, G.; Smith, D.; Sammond, D.; Tang, T.P.; Rhodes, S.P.; et al. Discovery of 7-tetrahydropyran-2-yl chromans: β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors that reduce amyloid β-protein (Aβ) in the central nervous system. J. Med. Chem. 2014, 57, 878–902. [Google Scholar] [CrossRef]
- Prati, F.; Bottegoni, G.; Bolognesi, M.L.; Cavalli, A. BACE-1 Inhibitors: From Recent Single-Target Molecules to Multitarget Compounds for Alzheimer’s Disease. J. Med. Chem. 2018, 61, 619–637. [Google Scholar] [CrossRef]
- Malamas, M.S.; Barnes, K.; Hui, Y.; Johnson, M.; Lovering, F.; Condon, J.; Fobare, W.; Solvibile, W.; Turner, J.; Hu, Y.; et al. Novel pyrrolyl 2-aminopyridines as potent and selective human β-secretase (BACE1) inhibitors. Bioorganic Med. Chem. Lett. 2010, 20, 2068–2073. [Google Scholar] [CrossRef]
- Hilpert, H.; Guba, W.; Woltering, T.J.; Wostl, W.; Pinard, E.; Mauser, H.; Mayweg, A.V.; Rogers-Evans, M.; Humm, R.; Krummenacher, D.; et al. β-secretase (BACE1) inhibitors with high in vivo efficacy suitable for clinical evaluation in Alzheimer’s disease. J. Med. Chem. 2013, 56, 3980–3995. [Google Scholar] [CrossRef]
- Epstein, O.; Bryan, M.C.; Cheng, A.C.; Derakhchan, K.; Dineen, T.A.; Hickman, D.; Hua, Z.; Human, J.B.; Kreiman, C.; Marx, I.E.; et al. Lead optimization and modulation of hERG activity in a series of aminooxazoline xanthene β-site amyloid precursor protein cleaving enzyme (BACE1) inhibitors. J. Med. Chem. 2014, 57, 9796–9810. [Google Scholar] [CrossRef]
- Cheng, Y.; Brown, J.; Judd, T.C.; Lopez, P.; Qian, W.; Powers, T.S.; Chen, J.J.; Bartberger, M.D.; Chen, K.; Dunn, R.T.; et al. An orally available BACE1 inhibitor that affords robust CNS Aβ reduction without cardiovascular liabilities. Acs Med. Chem. Lett. 2015, 6, 210–215. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. A Safety Study of LY2811376 Single Doses in Healthy Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT00838084 (accessed on 18 March 2019).
- May, P.C.; Dean, R.A.; Lowe, S.L.; Martenyi, F.; Sheehan, S.M.; Boggs, L.N.; Monk, S.A.; Mathes, B.M.; Mergott, D.J.; Watson, B.M.; et al. Robust Central Reduction of Amyloid-β in Humans with an Orally Available, Non-Peptidic -Secretase Inhibitor. J. Neurosci. 2011, 31, 16507–16516. [Google Scholar] [CrossRef]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Study of LY2886721 in Mild Cognitive Impairment Due to Alzheimer’s Disease or Mild Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01561430 (accessed on 18 March 2019).
- PRNewswire. Lilly Voluntarily Terminates Phase II Study for LY2886721, a Beta Secretase Inhibitor, Being Investigated as a Treatment for Alzheimer’s Disease. 2013. Available online: https://www.prnewswire.com/news-releases/lilly-voluntarily-terminates-phase-ii-study-for-ly2886721-a-beta-secretase-inhibitor-being-investigated-as-a-treatment-for-alzheimers-disease-211452171.html (accessed on 5 January 2019).
- ALZFORUM. RG7129. Available online: https://www.alzforum.org/therapeutics/rg7129 (accessed on 5 January 2019).
- Jacobsen, H.; Ozmen, L.; Caruso, A.; Narquizian, R.; Hilpert, H.; Jacobsen, B.; Terwel, D.; Tanghe, A.; Bohrmann, B. Combined Treatment with a BACE Inhibitor and Anti-Aβ Antibody Gantenerumab Enhances Amyloid Reduction in APP London Mice. J. Neurosci. 2014, 34, 11621–11630. [Google Scholar] [CrossRef]
- Hills, I.D.; Katharine Holloway, M.; de León, P.; Nomland, A.; Zhu, H.; Rajapakse, H.; Allison, T.J.; Munshi, S.K.; Colussi, D.; Pietrak, B.L.; et al. A conformational constraint improves a β-secretase inhibitor but for an unexpected reason. Bioorganic Med. Chem. Lett. 2009, 19, 4993–4995. [Google Scholar] [CrossRef]
- Malamas, M.S.; Erdei, J.; Gunawan, I.; Barnes, K.; Johnson, M.; Yu, H.; Turner, J.; Yun, H.; Wagner, E.; Fan, K.; et al. Aminoimidazoles as potent and selective human β-secretase (BACE1) inhibitors. J. Med. Chem. 2009, 52, 6314–6323. [Google Scholar] [CrossRef]
- Swahn, B.M.; Holenz, J.; Kihlström, J.; Kolmodin, K.; Lindström, J.; Plobeck, N.; Rotticci, D.; Sehgelmeble, F.; Sundström, M.; Fälting, J.; et al. Aminoimidazoles as BACE-1 inhibitors: The challenge to achieve in vivo brain efficacy. Bioorganic Med. Chem. Lett. 2012, 22, 1854–1859. [Google Scholar] [CrossRef]
- AstraZeneca. AstraZeneca and Lilly Announce Alliance to Develop and Commercialise BACE Inhibitor AZD3293 for Alzheimer’s Disease. 2014. Available online: https://www.astrazeneca.com/media-centre/press-releases/2014/astrazeneca-lilly-bace-inhibitor-azd3293-alzheimers-disease-16092014.html#! (accessed on 15 February 2019).
- Cebers, G.; Alexander, R.C.; Haeberlein, S.B.; Han, D.; Goldwater, R.; Ereshefsky, L.; Olsson, T.; Ye, N.; Rosen, L.; Russell, M.; et al. AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 55, 1039–1053. [Google Scholar] [CrossRef]
- ALZFORUM. Lanabecestat. Available online: https://www.alzforum.org/therapeutics/azd3293 (accessed on 5 January 2019).
- Menting, K.W.; Claassen, J.A.H.R. β-secretase inhibitor; a promising novel therapeutic drug in Alzheimer’s Disease. Front. Aging Neurosci. 2014, 6, 165. [Google Scholar] [CrossRef]
- Scott, J.D.; Li, S.W.; Brunskill, A.P.J.; Chen, X.; Cox, K.; Cumming, J.N.; Forman, M.; Gilbert, E.J.; Hodgson, R.A.; Hyde, L.A.; et al. Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)-A β-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2016, 59, 10435–10450. [Google Scholar] [CrossRef]
- Rochin, L.; Hurbain, I.; Serneels, L.; Fort, C.; Watt, B.; Leblanc, P.; Marks, M.S.; De Strooper, B.; Raposo, G.; van Niel, G. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10658–10663. [Google Scholar] [CrossRef] [Green Version]
- ALZFORUM. Verubecestat. Available online: https://www.alzforum.org/therapeutics/verubecestat (accessed on 5 January 2019).
- Merck. Merck Announces Discontinuation of APECS Study Evaluating Verubecestat (MK-8931) for the Treatment of People with Prodromal Alzheimer’s Disease. Available online: https://investors.merck.com/news/press-release-details/2018/Merck-Announces-Discontinuation-of-APECS-Study-Evaluating-Verubecestat-MK-8931-for-the-Treatment-of-People-with-Prodromal-Alzheimers-Disease/default.aspx (accessed on 13 March 2019).
- ALZFORUM. Bump in the Road or Disaster? BACE Inhibitors Worsen Cognition. Available online: https://www.alzforum.org/news/conference-coverage/bump-road-or-disaster-bace-inhibitors-worsen-cognition (accessed on 13 March 2019).






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure |
|---|---|
| Tacrine |  |
| Donepezil |  |
| Rivastigmine |  |
| Galantamine |  |
| Memantine |  |
| Phase (Clinical Trial(s)) | Agent | Mechanism of Action | Sponsor |
|---|---|---|---|
| I, III | Aducanumab | Monoclonal antibody | Biogen |
| III | Albumin + immunoglobulin | Polyclonal antibody | Grifols |
| I, II, III | Crenezumab | Monoclonal antibody | Roche/Genentech |
| II, III | Gantenerumab | Monoclonal antibody | Roche |
| III | Solanezumab | Monoclonal antibody | Washington University, Eli Lilly, Roche, NIA, Alzheimer’s Association, ATRI (Alzheimer’s Therapeutic Research Institute) |
| II, III | CAD106 | Amyloid vaccine | Novartis, Amgen, NIA, |
| II | Sargramostim (GM-CSF) | Immunostimulator | University of Colorado, Denver, The Dana Foundation |
| II | BAN2401 | Monoclonal antibody | Eisai |
| II | UB-311 | Monoclonal antibody | United Neuroscience |
| I, II | LY3002813 | Monoclonal antibody | Eli Lilly and Company |
| I | LY3303560 | Monoclonal antibody | Eli Lilly and Company |
| I | Lu AF20513 | Polyclonal antibody | H. Lundbeck A/S |
| I | KHK6640 | Monoclonal antibody | Kyowa Hakko Kirin Pharma |
| Phase Clinical Trial(s) | Agent | Mechanism of Action | Sponsor |
|---|---|---|---|
| I | NGP 555 | GSM | NeuroGenetic Pharmaceuticals |
| II | ID1201 | Phosphatidylinositol 3-kinase/Akt pathway activation | II Dong Pharmaceutical Co |
| II | Nilotinib | Tyrosine kinase inhibitor | Georgetown University |
| III | CNP520 | (γ-secretase modulator) | Alzheimer’s Association |
| III | ALZT-OP1a (cromolyn)+ ALZT-OP1b (ibuprofen) | BACE1 inhibitor | AZTherapies |
| III | Sodium Oligo-mannurarate (GV-971) | Increases amyloid clearance | Shanghai Green Valley |
| III | TTP488 (Azeliragon) | RAGE1) (Receptor for advanced glycation end products) antagonist | vTv Therapeutics |
| II, III | JNJ-54861911 | BACE1 inhibitor | Janssen |
| II, III | E2609 (Elenbecestat) | BACE1 inhibitor | Eisai, Biogen |
| II | LY3202626 | BACE1 inhibitor | Eli Lilly |
| II | Atomoxetine | Adrenergic uptake inhibitor, SNRI | Emory University, NIA |
| II | AZD0530 (Saracatinib) | Kinase inhibitor | Yale University, ATRI, |
| II | CT1812 | Sigma-2 receptor competitive inhibitor2) | Cognition Therapeutics |
| II | Posiphen | Selective inhibitor of APP production | QR Pharma, ADCS |
| II | Valacyclovir | Antiviral agent 4) | Umea University |
| III | AZD3293 (LY3314814) | BACE1 inhibitor | AstraZeneca, Eli Lilly |
| Amino Acid Residues | Amino Acid Residues | ||
|---|---|---|---|
| S1 | Leu30, Asp32, Tyr71, Leu119, Gln73, Phe108 | S1′ | Val31, Tyr71, Thr72, Asp228 |
| S2 | Asn233, Arg235, Ser325 | S2′ | Ser35, Val69, Pro70, Tyr198 |
| S3 | Leu133, Ile110, Ser229 | S3′ | Arg128, Tyr198 |
| S4 | Gln73, Thr232, Arg307 | S4′ | Glu125, Ile126, Tyr197, Tyr198 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia, M.A.; Sousa, E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals 2019, 12, 41. https://doi.org/10.3390/ph12010041
Maia MA, Sousa E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals. 2019; 12(1):41. https://doi.org/10.3390/ph12010041
Chicago/Turabian StyleMaia, Miguel A., and Emília Sousa. 2019. "BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease" Pharmaceuticals 12, no. 1: 41. https://doi.org/10.3390/ph12010041
APA StyleMaia, M. A., & Sousa, E. (2019). BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals, 12(1), 41. https://doi.org/10.3390/ph12010041