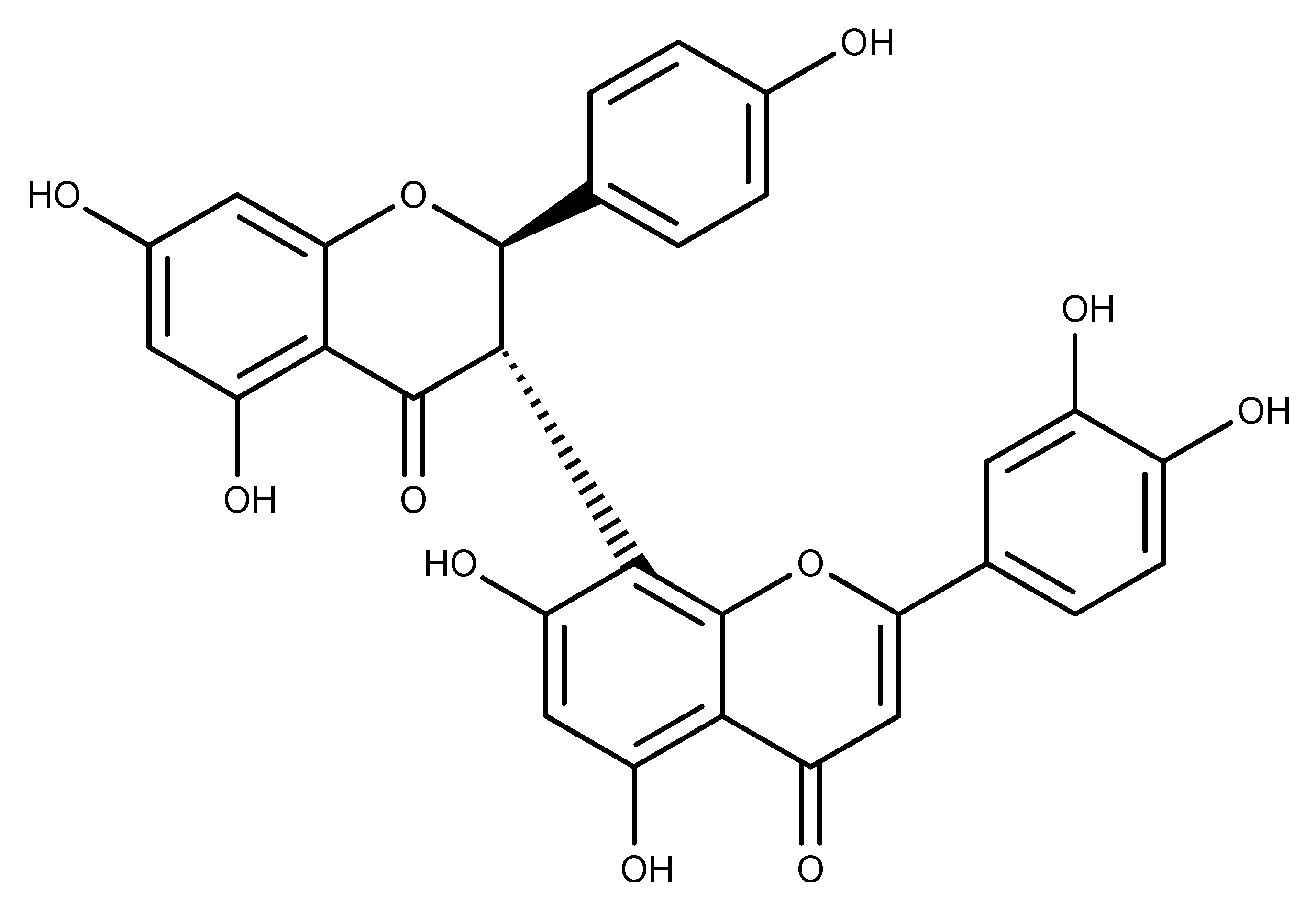
Insights into the Molecular Mechanisms of Eg5 Inhibition by (+)-Morelloflavone




Abstract
:1. Introduction
2. Results and Discussion
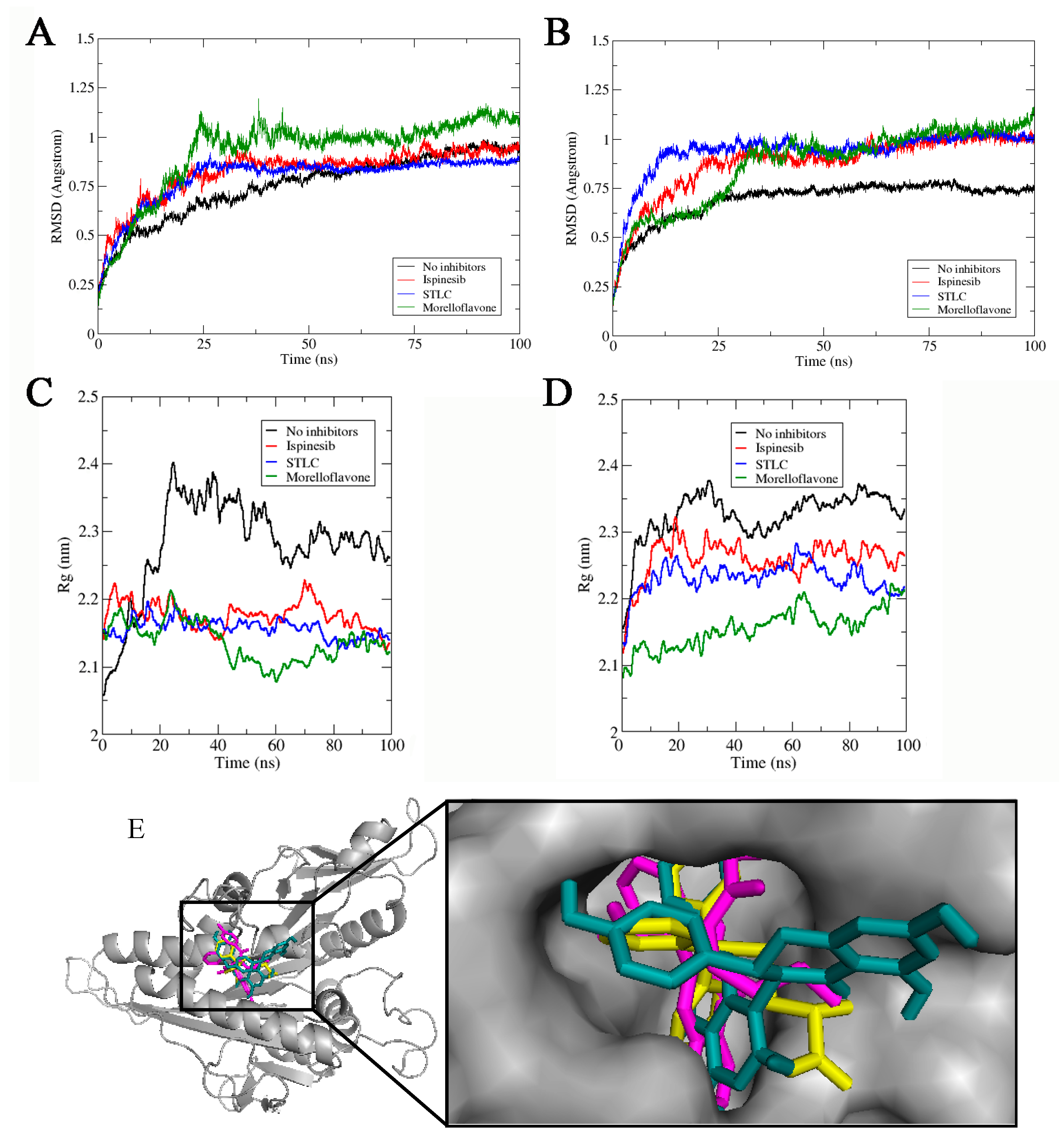
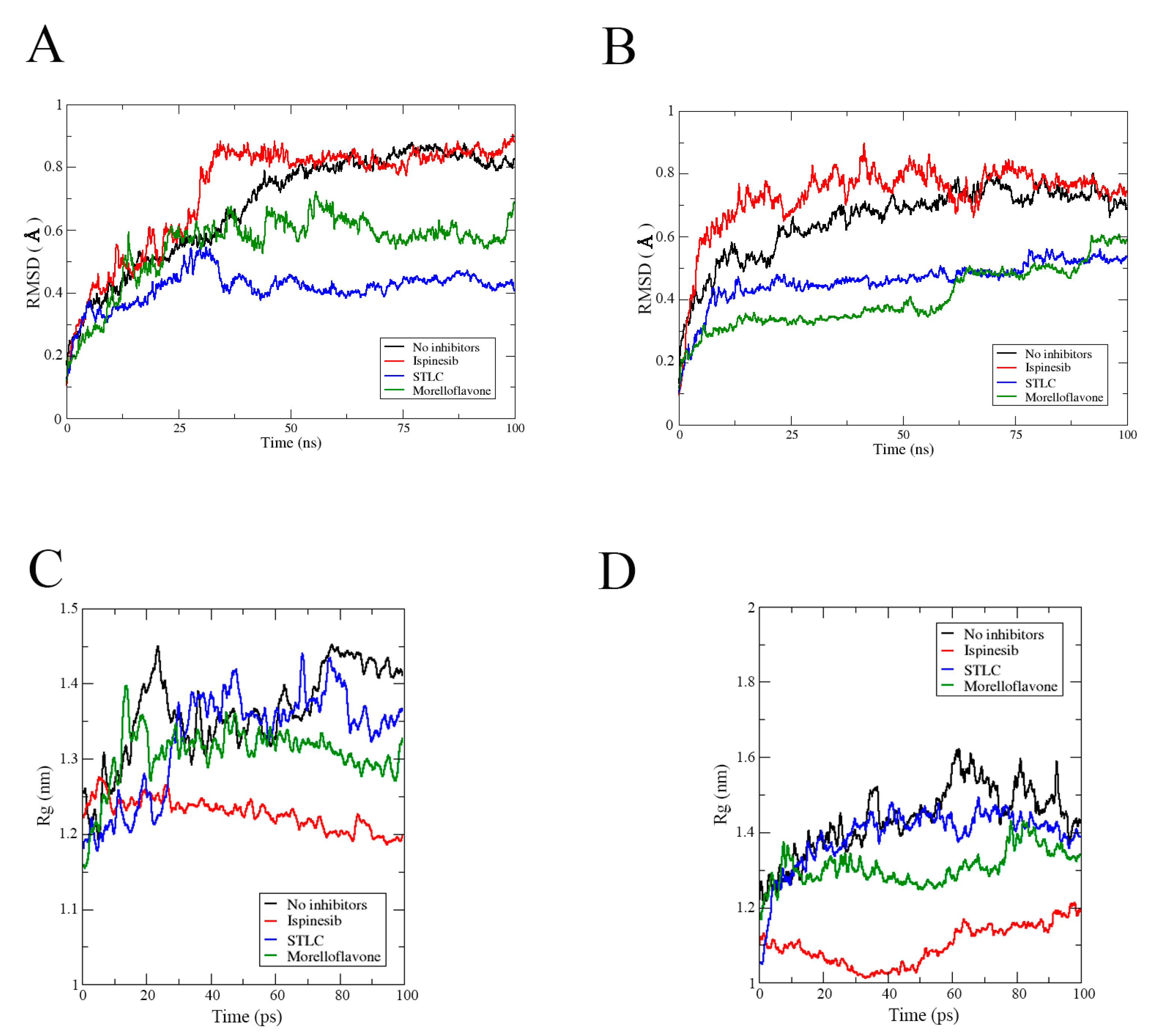
2.1. MF Exhibits Stability in Complex with Eg5 at the Allosteric Pocket
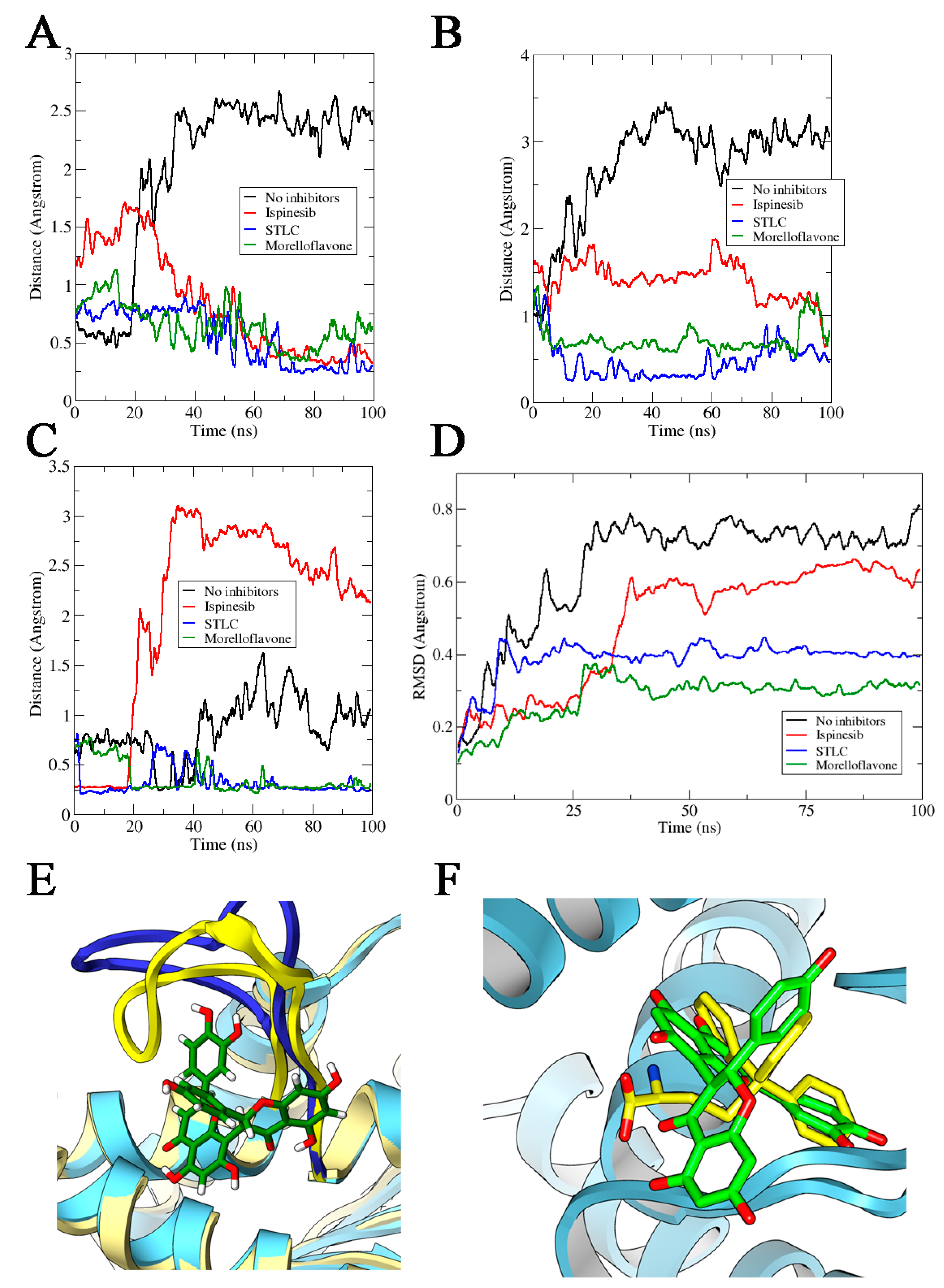
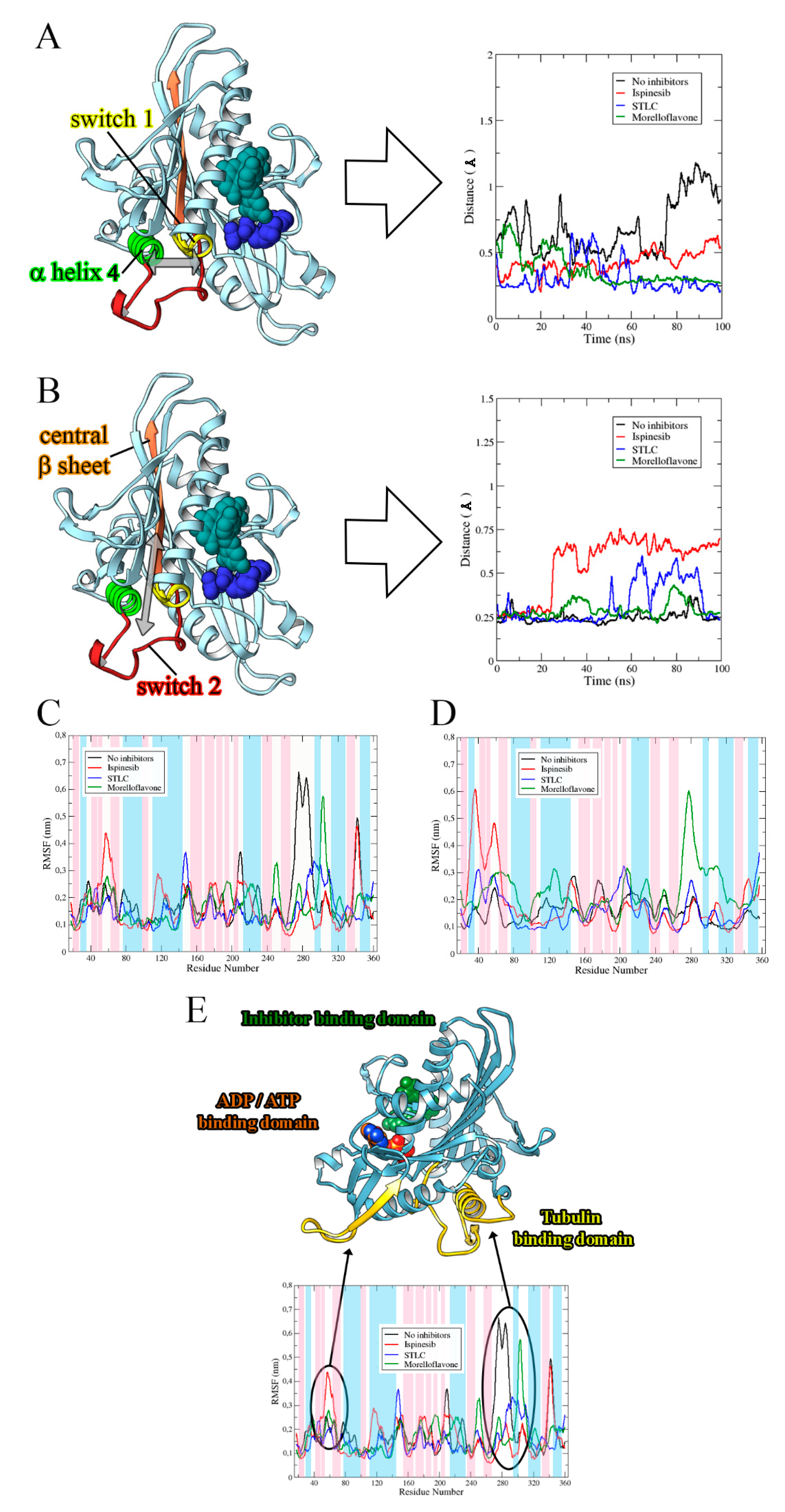
2.2. MF Induces Eg5-Loop5/α2/α3 Pocket Closure in a Manner Comparable to STLC
2.3. MF Induces Compactness and Stabilizes the Eg5 Allosteric Pocket
2.4. Binding of MF on the Allosteric Site Alters the Eg5 Structural Conformation
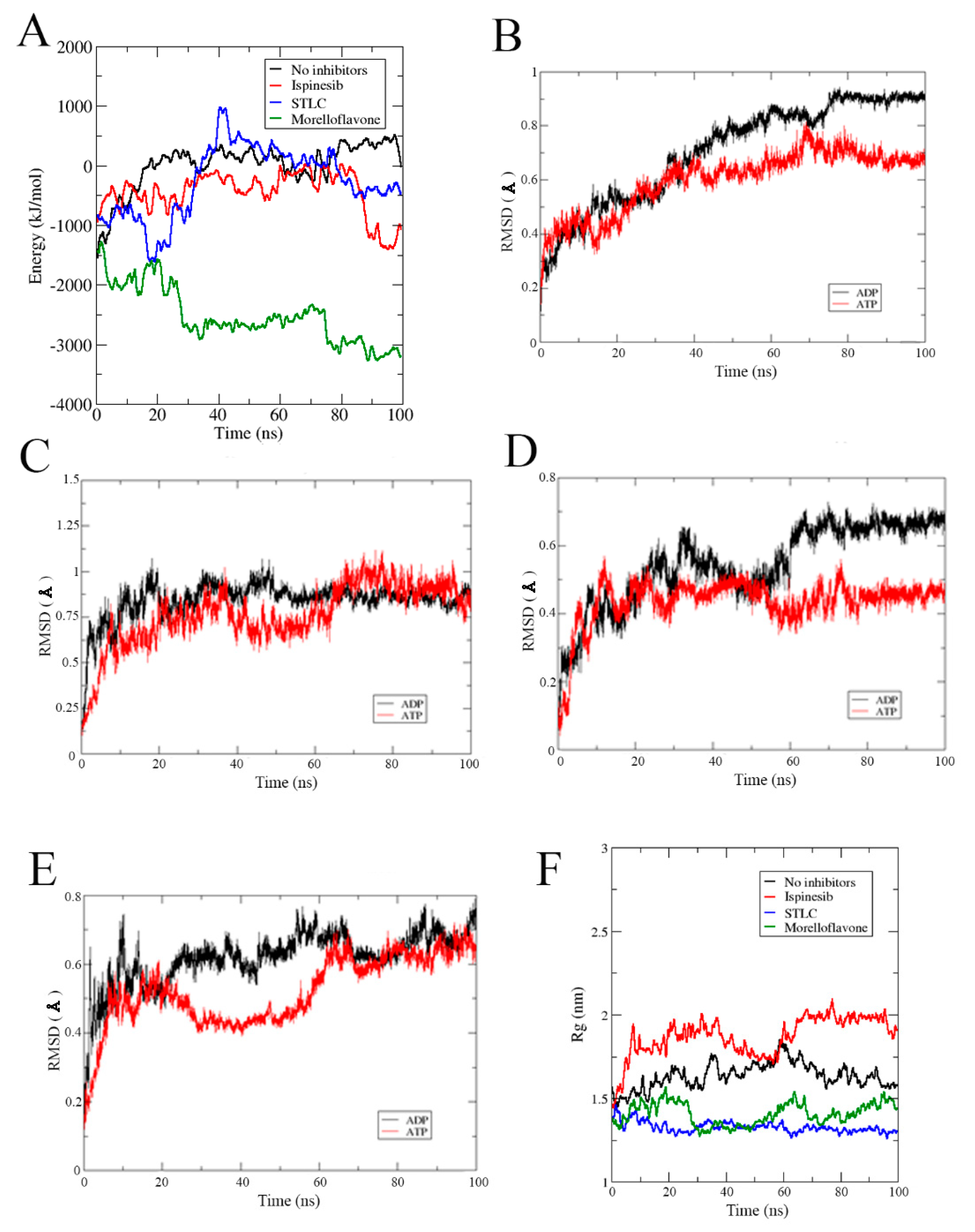
2.5. Binding Free Energy Estimation for Eg5-Inhibitor Complexes
- Bound state (windows 0–5 for Ispinesib, 0–7 for MF, and 0–8 for STLC) where the inhibitors are tightly bound to Eg5 via their numerous charge contacts. This effectively restricts the inhibitors’ rotational freedoms such that similar behavior is observed across all trajectories.
- Transition state (windows 5–10 for Ispinesib, 7–10 for MF, and 8–10 for STLC) where inhibitors must undergo some rotation to break out. In this region, the interactions between inhibitors and Eg5 are gradually broken. This corresponds to increasing rotational freedom in the molecules.
- Electrostatic region (windows 10–14 for Ispinesib, 10–14 for MF, and 10–12 for STLC) where all direct contacts have been severed and residual electrostatic interactions have been screened by incoming water molecules. Under weak electrostatic attractions, the inhibitor molecules are more or less rotationally free.
2.6. MF Alters the Affinity of Nucleotides to the Active Site of Eg5
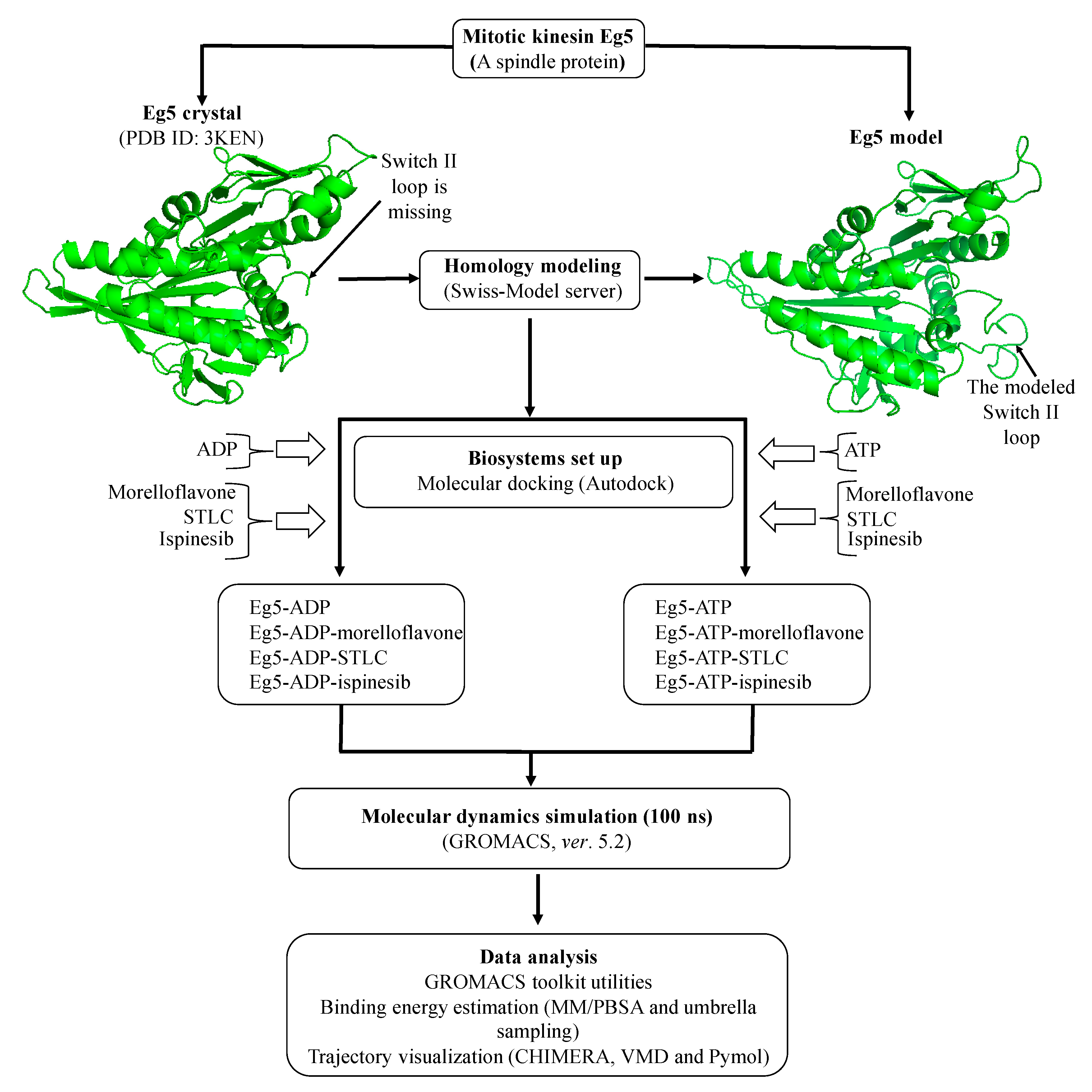
3. Computational Methods
3.1. Starting Structures
3.2. Biosystems Setup
3.3. MD Simulation
3.4. Umbrella Sampling and Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mandelkow, E.; Mandelkow, E.M. Kinesin motors and disease. Trends Cell Biol. 2002, 12, 585–591. [Google Scholar] [CrossRef]
- Hirokawa, N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998, 279, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Endow, S.A. Microtubule motors in spindle and chromosome motility. Eur. J. Biochem. 1999, 262, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Wordeman, L. How kinesin motor proteins drive mitotic spindle function: Lessons from molecular assays. Semin. Cell Dev. Biol. 2010, 21, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Sudakin, V.; Yen, T.J. Targeting mitosis for anti-cancer therapy. BioDrugs 2007, 21, 225–233. [Google Scholar] [CrossRef]
- Sherr, C.J.; Bartek, J. Cell cycle-targeted cancer therapies. Annu. Rev. Cancer Biol. 2017, 1, 41–57. [Google Scholar] [CrossRef]
- Salmela, A.L.; Kallio, M.J. Mitosis as an anti-cancer drug target. Chromosoma 2013, 122, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Ferenz, N.P.; Gable, A.; Wadsworth, P. Mitotic functions of kinesin-5. Semin. Cell Dev. Biol. 2010, 21, 255–259. [Google Scholar] [CrossRef]
- Waitzman, J.S.; Rice, S.E. Mechanism and regulation of kinesin-5, an essential motor for the mitotic spindle. Biol. Cell 2014, 106, 1–12. [Google Scholar] [CrossRef]
- Kapoor, T.M.; Mayer, T.U.; Coughlin, M.L.; Mitchison, T.J. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 2000, 150, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, X.; Du, M.; Chen, X.; Ning, X.; Chen, H.; Wang, S.; Liu, J.; Liu, Z.; Li, R.; et al. Eg5 inhibitor YL001 induces mitotic arrest and inhibits tumor proliferation. Oncotarget 2017, 8, 42510–42524. [Google Scholar] [CrossRef] [PubMed]
- Skoufias, D.A.; DeBonis, S.; Saoudi, Y.; Lebeau, L.; Crevel, I.; Cross, R.; Wade, R.H.; Hackney, D.; Kozielski, F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 2006, 281, 17559–17569. [Google Scholar] [CrossRef] [PubMed]
- Lad, L.; Luo, L.; Carson, J.D.; Wood, K.W.; Hartman, J.J.; Copeland, R.A.; Sakowicz, R. Mechanism of inhibition of human KSP by ispinesib. Biochemistry 2008, 47, 3576–3585. [Google Scholar] [CrossRef] [PubMed]
- Schiemann, K.; Finsinger, D.; Zenke, F.; Amendt, C.; Knöchel, T.; Bruge, D.; Buchstaller, H.P.; Emde, U.; Stähle, W.; Anzali, S. The discovery and optimization of hexahydro-2H-pyrano[3,2-c]quinolines (HHPQs) as potent and selective inhibitors of the mitotic kinesin-5. Bioorg. Med. Chem. Lett. 2010, 20, 1491–1495. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Anthony, N.G.; Mackay, S.P.; Kozielski, F. Mitotic kinesin Eg5 overcomes inhibition to the phase I/II clinical candidate SB743921 by an allosteric resistance mechanism. J. Med. Chem. 2013, 56, 6317–6329. [Google Scholar] [CrossRef] [PubMed]
- Rickert, K.W.; Schaber, M.; Torrent, M.; Neilson, L.A.; Tasber, E.S.; Garbaccio, R.; Coleman, P.J.; Harvey, D.; Zhang, Y.; Yang, Y.; et al. Discovery and biochemical characterization of selective ATP competitive inhibitors of the human mitotic kinesin KSP. Arch. Biochem. Biophys. 2008, 469, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.A.; Adams, N.D.; Auger, K.R.; Burgess, J.L.; Carson, J.D.; Chaudhari, A.M.; Copeland, R.A.; Diamond, M.A.; Donatelli, C.A.; Duffy, K.J.; et al. Novel ATP-competitive kinesin spindle protein inhibitors. J. Med. Chem. 2007, 50, 4939–4952. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Lowndes, N.F.; Eriksson, L.A. Analysis of Biphenyl-Type Inhibitors Targeting the Eg5 α4/α6 Allosteric Pocket. ACS Omega 2017, 2, 1836–1849. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Parrish, C.A.; Nevins, N.; McNulty, D.E.; Chaudhari, A.M.; Carson, J.D.; Sudakin, V.; Shaw, A.N.; Lehr, R.; Zhao, H.; et al. ATP-competitive inhibitors of the mitotic kinesin KSP that function via an allosteric mechanism. Nat. Chem. Biol. 2007, 3, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chen, Y.; Wang, X.; You, Q. Docking studies on kinesin spindle protein inhibitors: An important cooperative “minor binding pocket” which increases the binding affinity significantly. J. Mol. Model. 2007, 13, 987e992. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Schüttelkopf, A.W.; Kozielski, F. The structure of the ternary Eg5-ADP-ispinesib complex. Acta Crystallogr. Biol. Crystallogr. 2012, 68, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Sardana, V.; Xu, B.; Homnick, C.; Halczenko, W.; Buser, C.A.; Schaber, M.; Hartman, G.D.; Huber, H.E.; Kuo, L.C. Inhibition of a mitotic motor protein: Where, how, and conformational consequences. J. Mol. Biol. 2004, 335, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Moores, C.A. Kinesin-5 mitotic motors: Is loop5 the on/off switch? Cell Cycle 2010, 9, 1286–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, S.; Skoufias, D.A.; Kozielski, F.; Pae, A.N. Receptor ligand interaction-based virtual screening for novel Eg5/kinesin spindle protein inhibitors. J. Med. Chem. 2012, 55, 2561–2573. [Google Scholar] [CrossRef]
- Ogo, N.; Ishikawa, Y.; Sawada, J.; Matsuno, K.; Hashimoto, A.; Asai, A. Structure-guided design of novel l-cysteine derivatives as potent KSP inhibitors. ACS Med. Chem. Lett. 2015, 6, 1004–1009. [Google Scholar] [CrossRef]
- Li, X.; Ai, H.; Sun, D.; Wu, T.; He, J.; Xu, Z.; Ding, L.; Wang, L. Anti-tumoral activity of native compound morelloflavone in glioma. Oncol. Lett. 2016, 12, 3373–3377. [Google Scholar] [CrossRef] [Green Version]
- Ogunwa, T.H.; Kenichi, T.; Kei, S.; Yuka, K.; Shinsaku, M.; Takayuki, M. Morelloflavone as novel inhibitor for mitotic kinesin Eg5. J. Biochem. 2019, in press. [Google Scholar] [CrossRef]
- Li, X.C.; Joshi, A.S.; Tan, B.; El Sohly, H.N.; Walker, L.A.; Zjawiony, J.K.; Ferreira, D. Absolute configuration, conformation, and chiral properties of flavanone-(3-8")-flavone biflavonoids from Rheedia Acuminata. Tetrahedron 2002, 58, 8709–8717. [Google Scholar] [CrossRef]
- Farrell, C.M.; Mackey, A.T.; Klumpp, L.M.; Gilbert, S.P. The role of ATP hydrolysis for kinesin processivity. J. Biol. Chem. 2002, 277, 17079–17087. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.J.; Kuo, I.F.; Hayashi, S.; Takada, S. Adenosine triphosphate hydrolysis mechanism in kinesin studied by combined quantum-mechanical/molecular-mechanical metadynamics simulations. J. Am. Chem. Soc. 2013, 135, 8908–8919. [Google Scholar] [CrossRef] [PubMed]
- Scarabelli, G.; Grant, B.J. Kinesin-5 allosteric inhibitors uncouple the dynamics of nucleotide, microtubule, and neck-linker binding sites. Biophys. J. 2014, 107, 2204–2213. [Google Scholar] [CrossRef]
- Kumaresan, J.; Kothai, T.; Lakshmi, B.S. In silico approaches towards understanding CALB using molecular dynamics simulation and docking. Mol. Simul. 2011, 37, 1053–1061. [Google Scholar] [CrossRef]
- Kaan, H.Y.; Major, J.; Tkocz, K.; Kozielski, F.; Rosenfeld, S.S. “Snapshots” of ispinesib-induced conformational changes in the mitotic kinesin Eg5. J. Biol. Chem. 2013, 288, 18588–18598. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.D.; Buckley, R.; Learman, S.; Richard, J.; Parke, C.; Worthylake, D.K.; Wojcik, E.J.; Walker, R.A.; Kim, S. Allosteric drug discrimination is coupled to mechanochemical changes in the kinesin-5 motor core. J. Biol. Chem. 2010, 285, 18650–18661. [Google Scholar] [CrossRef]
- Kaan, H.Y.; Ulaganathan, V.; Hackney, D.D.; Kozielski, F. An allosteric transition trapped in an intermediate state of a new kinesin-inhibitor complex. Biochem. J. 2009, 425, 55–60. [Google Scholar] [CrossRef]
- Garcia-Saez, I.; DeBonis, S.; Lopez, R.; Trucco, F.; Rousseau, B.; Thuery, P.; Kozielski, F. Structure of human Eg5 in complex with a new monastrol-based inhibitor bound in the R configuration. J. Biol. Chem. 2007, 282, 9740–9747. [Google Scholar] [CrossRef]
- Zhang, W. Exploring the intermediate states of ADP-ATP exchange: A simulation study on Eg5. J. Phys. Chem. 2011, 115, 784–795. [Google Scholar] [CrossRef]
- Nagarajan, S.; Sakkiah, S. Exploring a potential allosteric inhibition mechanism in the motor domain of human Eg5. J. Biomol. Struct. Dyn. 2018, 13, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Parke, C.L.; Wojcik, E.J.; Kim, S.; Worthylake, D.K. ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J. Biol. Chem. 2010, 285, 5859–5867. [Google Scholar] [CrossRef] [PubMed]
- Behnke-Parks, W.M.; Vendome, J.; Honig, B.; Maliga, Z.; Moores, C.; Rosenfeld, S.S. Loop L5 acts as a conformational latch in the mitotic kinesin Eg5. J. Biol. Chem. 2011, 286, 5242–5253. [Google Scholar] [CrossRef]
- Luo, L.; Carson, J.D.; Dhanak, D.; Jackson, J.R.; Huang, P.S.; Lee, Y.; Sakowicz, R.; Copeland, R.A. Mechanism of inhibition of human KSP by monastrol: Insights from kinetic analysis and the effect of ionic strength on KSP inhibition. Biochemistry 2004, 43, 15258–15266. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Kang, Y.J.; Gayek, A.S.; Youyen, W.; Tüzel, E.; Ohi, R.; Hancock, W.O. Eg5 inhibitors have contrasting effects on microtubule stability and metaphase spindle integrity. ACS Chem. Biol. 2017, 12, 1038–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Kumar, R. Open source drug discovery consortium, Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- MarvinSketch (version 15.7.27). Calculation Module Developed by ChemAxon. Available online: http://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 6 April 2019).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; Lepore, R.; Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Meagher, K.L.; Redman, L.T.; Carlson, H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24, 1016–1025. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D. Solvation and conformation of methanol in water. Am. Chem. Soc. 1983, 105, 1407–1413. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Jakalian, A.; David, B.J.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline side chain dynamics. Proteins 2014, 82, 195–215. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, J.J.; Kim, S.; Lee, S.C.; Han, J.; Heu, W.; Park, K.; Kim, H.J.; Cheong, H.K.; Kim, D.; Kim, H.S.; Lee, K.W. Dissecting the critical factors for thermodynamic stability of modular proteins using molecular modeling approach. PLoS ONE 2014, 9, e98243. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Laudadio, E.; Mobbili, G.; Minnelli, C.; Massaccesi, L.; Galeazzi, R. Salts influence cathechins and flavonoids encapsulation in liposomes: A molecular dynamics investigation. Mol. Inform. 2017, 36, 1700059. [Google Scholar] [CrossRef]
- Mangiaterra, G.; Laudadio, E.; Cometti, M.; Mobbili, G.; Minnelli, C.; Massaccesi, L.; Biavasco, F.; Citterio, B.; Galeazzi, R. Inhibitors of multidrug efflux pumps of Pseudomonas aeruginosa from natural sources: An in silico high-throughput virtual screening and in vitro validation. Med. Chem. Res. 2017, 26, 414. [Google Scholar] [CrossRef]
- Galeazzi, R.; Mobbili, G.; Laudadio, E.; Minnelli, C.; Amici, A.; Massaccesi, L. Liposomial formulations for an efficient encapsulation Epigallocatechin-3-gallate: An in silico/experimental approach. Molecules 2018, 23, 441. [Google Scholar]
- Nosè, S. A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Bevan, D.R. Assessing the stability of Alzheimer’s amyloid protofibrils using molecular dynamics. J. Phys. Chem. 2010, 114, 1652–1660. [Google Scholar] [CrossRef]
- Grossfield, A. An implementation of WHAM: The Weighted Histogram Analysis Method, version 2.0.9. Available online: https://project-us.mimecast.com/s/uq-tCXD7MEUXy7QnktMii4N?domain=membranee.urmc.rochester.edu (accessed on 6 April 2019).
- Tilio, M.; Gambini, V.; Wang, J.; Garulli, C.; Kalogris, C.; Andreani, C.; Bartolacci, C.; Elexpuru Zabaleta, M.; Pietrella, L.; Hysi, A.; et al. Irreversible inhibition of Δ16HER2 is necessary to suppress Δ16HER2-positive breast carcinomas resistant to Lapatinib. Cancer Lett. 2016, 10, 76–84. [Google Scholar]
- Fedeli, D.; Montani, M.; Bordoni, L.; Galeazzi, R.; Nasuti, C.; Correia-Sá, L.; Domingues, V.F.; Jayant, M.; Brahmachari, V.; Massaccesi, L.; et al. In vivo and in silico studies to identify mechanisms associated with nurr1 modulation following early life exposure to permethrin in rats. Neuroscience 2017, 340, 411–423. [Google Scholar] [CrossRef]
- Gabbianelli, R.; Carloni, M.; Marmocchi, F.; Nasuti, C.; Fedeli, D.; Laudadio, E.; Massaccesi, L.; Galeazzi, R. Permethrin and its metabolites affects Cu/Zn Superoxide conformation: Fluorescence and in silico evidences. Mol. BioSyst. 2015, 11, 208–217. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Turner, P.J. XMGRACE, Version 5.1.21; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Galeazzi, R.; Bruni, P.; Crucianelli, E.; Laudadio, E.; Marini, M.; Massaccesi, L.; Mobbili, G.; Pisani, M. Liposome-based gene delivery systems containing a steroid derivative: Computational and small angle X-ray diffraction study. RSC Adv. 2015, 5, 54070. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy (kJ/mol) | Eg5-Ligand Complexes | ||
|---|---|---|---|
| EG5+ADP+Ispinesib | EG5+ADP+STLC | EG5+ADP+MF | |
| Van der Waals | −4.5 ± 8.50 | −20.3 ± 9.0 | −20.7 ± 8.7 |
| Electrostatic | −103.6 ± 11.0 | −158.7 ± 10.7 | −156.1 ± 11.1 |
| Polar solvation | −65.1 ± 9.1 | −36.1 ± 7.5 | −40.1 ± 7.7 |
| Non-polar solvation | 126.9 ± 34.2 | 126.1 ± 34.3 | 123.2 ± 33.4 |
| Binding energy | −96.31 ± 9.5 | −89.0 ± 8.9 | −93.7 ± 9.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogunwa, T.H.; Laudadio, E.; Galeazzi, R.; Miyanishi, T. Insights into the Molecular Mechanisms of Eg5 Inhibition by (+)-Morelloflavone. Pharmaceuticals 2019, 12, 58. https://doi.org/10.3390/ph12020058
Ogunwa TH, Laudadio E, Galeazzi R, Miyanishi T. Insights into the Molecular Mechanisms of Eg5 Inhibition by (+)-Morelloflavone. Pharmaceuticals. 2019; 12(2):58. https://doi.org/10.3390/ph12020058
Chicago/Turabian StyleOgunwa, Tomisin Happy, Emiliano Laudadio, Roberta Galeazzi, and Takayuki Miyanishi. 2019. "Insights into the Molecular Mechanisms of Eg5 Inhibition by (+)-Morelloflavone" Pharmaceuticals 12, no. 2: 58. https://doi.org/10.3390/ph12020058
APA StyleOgunwa, T. H., Laudadio, E., Galeazzi, R., & Miyanishi, T. (2019). Insights into the Molecular Mechanisms of Eg5 Inhibition by (+)-Morelloflavone. Pharmaceuticals, 12(2), 58. https://doi.org/10.3390/ph12020058