CDK8-Novel Therapeutic Opportunities

{kind=link}
{kind=link}
{kind=link}
Abstract
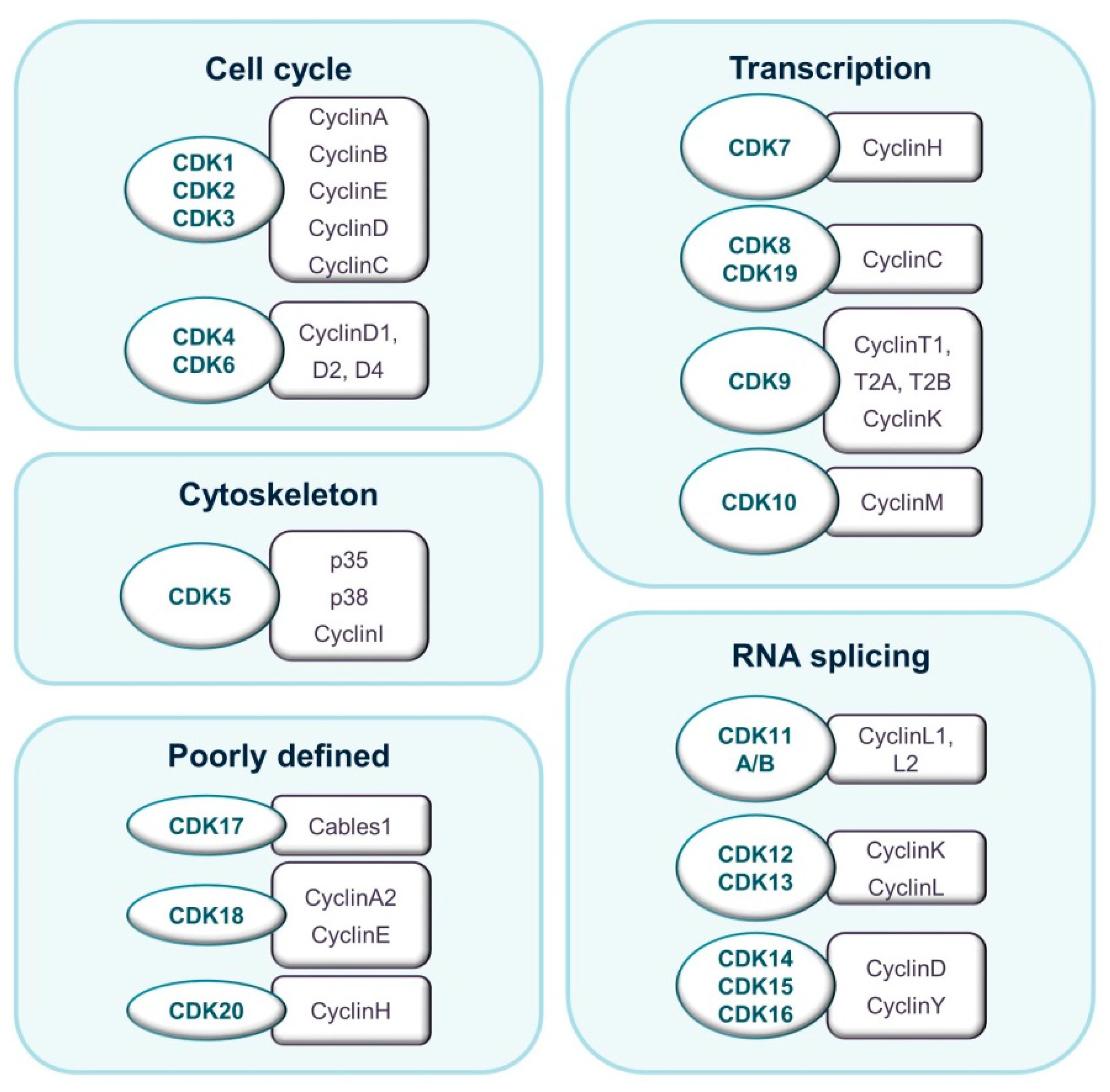
:1. Introduction
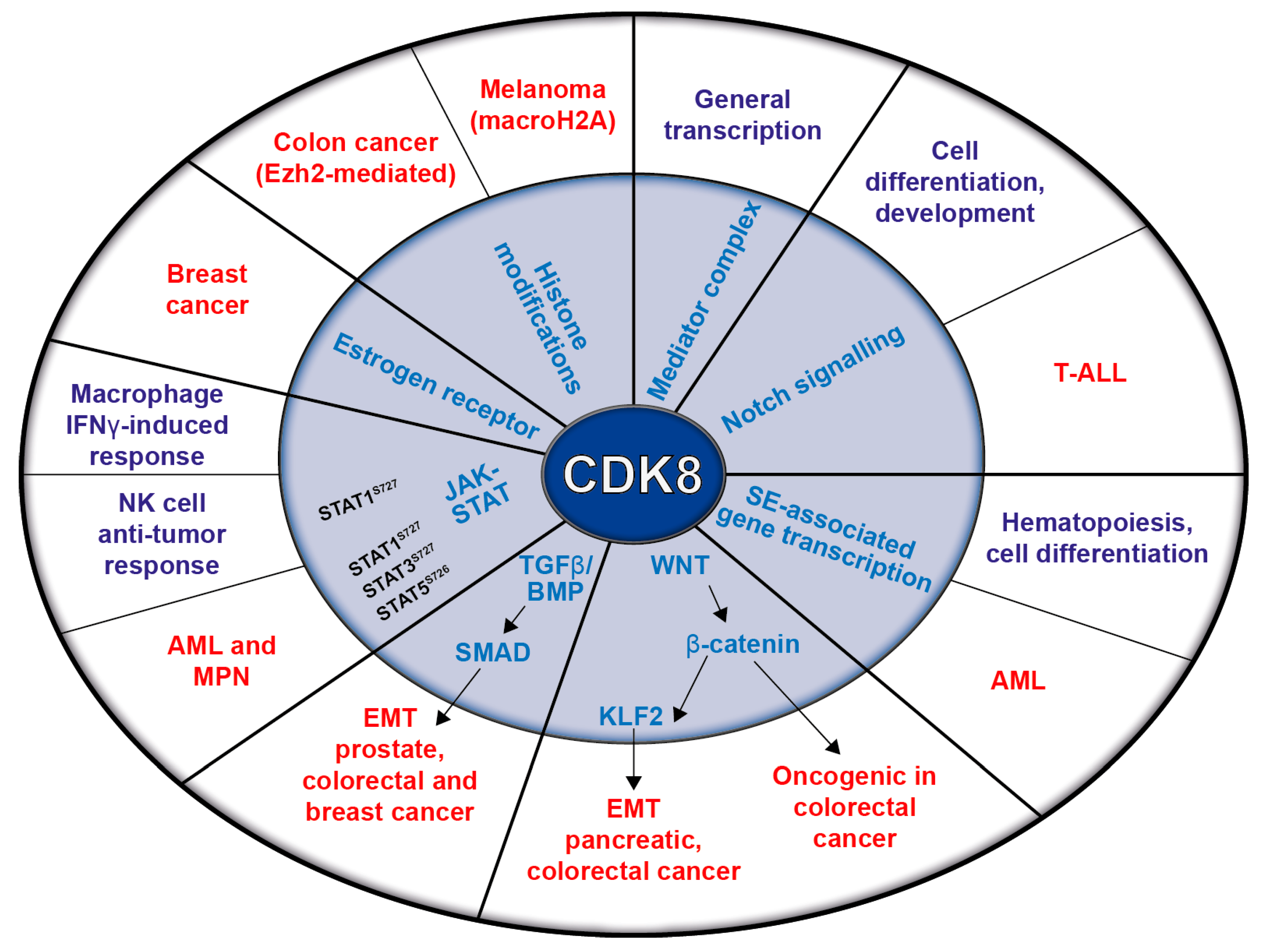
2. CDK8 as a Regulator of Transcription
2.1. CDK8 and the Mediator throughout Evolution
2.2. CDK8 in the Regulation of Gene Expression
2.3. CDK8 as A Promoter of Transcription Factor Activity and Degradation
3. CDK8 in Solid Tumors
CDK8 Triggers EMT and Invasiveness
4. CDK8 and Leukemia
5. CDK8 and the Immune System
6. CDK8 in Cancer Metabolism
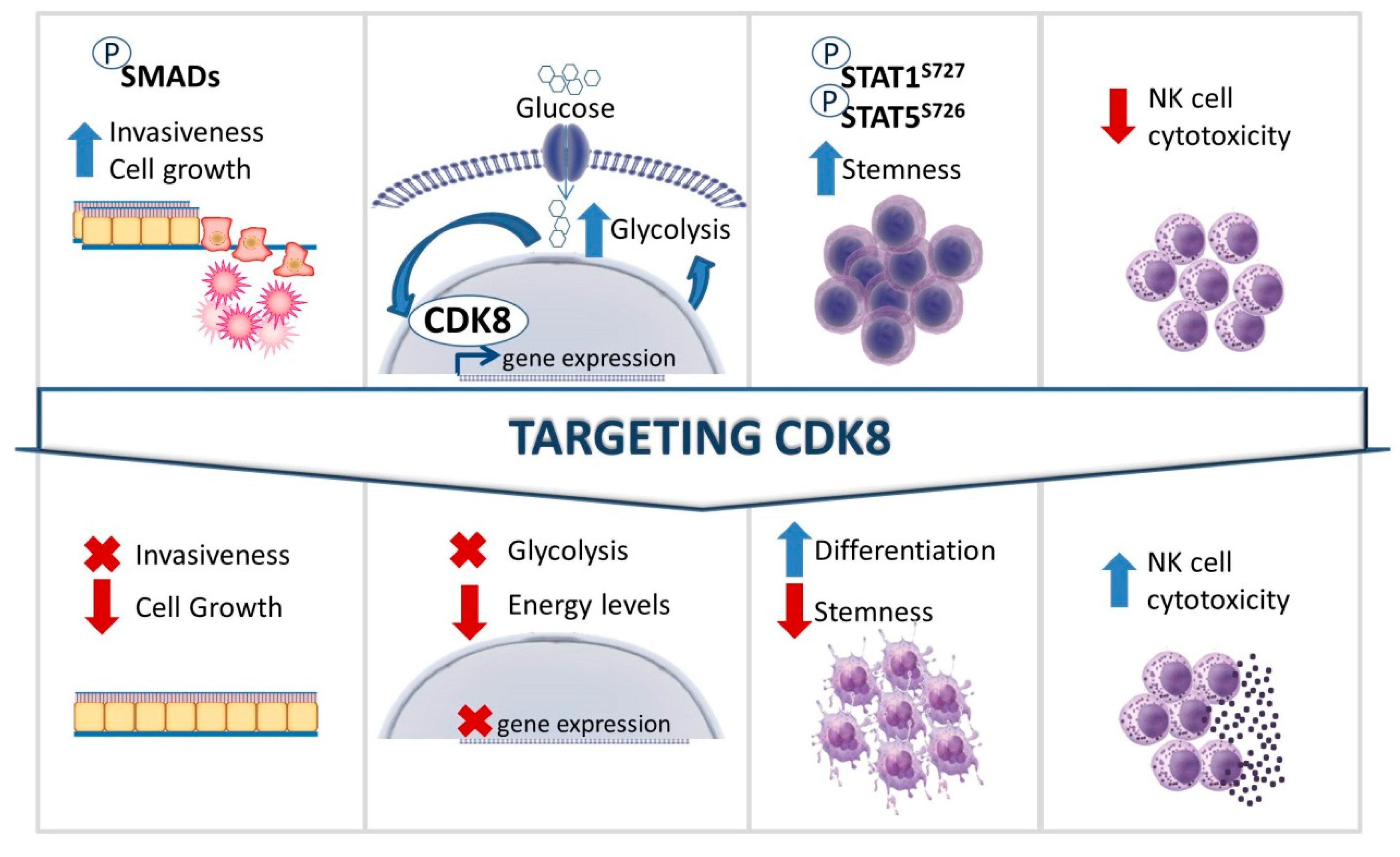
7. Potential Therapeutic Benefits of Targeting CDK8 in Cancer
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Russell, P.; Nurse, P. Schizosaccharomyces pombe and Saccharomyces cerevisiae: a look at yeasts divided. Cell 1986, 45, 781–782. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Manning, G.; Whyte, D.; Martinez, R.; Hunter, T.; Sudarsanam, S.; Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; et al. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol. Biol. 2014, 14, 10. [Google Scholar] [CrossRef]
- Hengartner, C.J.; Myer, V.E.; Liao, S.M.; Wilson, C.J.; Koh, S.S.; Young, R.A. Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol. Cell 1998, 2, 43–53. [Google Scholar] [CrossRef]
- Chi, Y.; Huddleston, M.J.; Zhang, X.; Young, R.A.; Annan, R.S.; Carr, S.A.; Deshaies, R.J. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 2001, 15, 1078–1092. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.; Goto, S.; Lund, K.; Hung, W.; Sadowski, I. Srb10/Cdk8 regulates yeast filamentous growth by phosphorylating the transcription factor Ste12. Nature 2003, 421, 187–190. [Google Scholar] [CrossRef]
- Liu, Y.; Kung, C.; Fishburn, J.; Ansari, A.Z.; Shokat, K.M.; Hahn, S. Two cyclin-dependent kinases promote RNA polymerase II transcription and formation of the scaffold complex. Mol. Cell. Biol. 2004, 24, 1721–1735. [Google Scholar] [CrossRef]
- Andrau, J.-C.; van de Pasch, L.; Lijnzaad, P.; Bijma, T.; Koerkamp, M.G.; van de Peppel, J.; Werner, M.; Holstege, F.C.P. Genome-Wide Location of the Coactivator Mediator: Binding without Activation and Transient Cdk8 Interaction on DNA. Mol. Cell 2006, 22, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wirén, M.; Sinha, I.; Rasmussen, N.N.; Linder, T.; Holmberg, S.; Ekwall, K.; Gustafsson, C.M. Genome-Wide Occupancy Profile of Mediator and the Srb8-11 Module Reveals Interactions with Coding Regions. Mol. Cell 2006, 22, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Howard, S.C.; Budovskaya, Y.V.; Rine, J.; Herman, P.K. The rye mutants identify a role for Ssn/Srb proteins of the RNA polymerase II holoenzyme during stationary phase entry in Saccharomyces cerevisiae. Genetics 2001, 157, 17–26. [Google Scholar] [PubMed]
- Loncle, N.; Boube, M.; Joulia, L.; Boschiero, C.; Werner, M.; Cribbs, D.L.; Bourbon, H.M. Distinct roles for Mediator Cdk8 module subunits in Drosophila development. EMBO J. 2007, 26, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.-W.; Hu, Y.; Chen, C.-L.; Xia, B.; Zirin, J.; Yuan, M.; Asara, J.M.; Rabinow, L.; Perrimon, N. The TORC1-Regulated CPA Complex Rewires an RNA Processing Network to Drive Autophagy and Metabolic Reprogramming. Cell Metab. 2018, 27, 1040–1054.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerling, T.; Kuuluvainen, E.; Mäkelä, T.P. Cdk8 is essential for preimplantation mouse development. Mol. Cell. Biol. 2007, 27, 6177–6182. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Soukup, T.M.; Liu, S.D.; Esensten, J.H.; De Sousa E Melo, F.; Yaylaoglu, M.; Warming, S.; Roose-Girma, M.; Firestein, R. Cdk8 deletion in the ApcMin murine tumour model represses EZH2 activity and accelerates tumourigenesis. J. Pathol. 2015, 237, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Akoulitchev, S.; Chuikov, S.; Reinberg, D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000, 407, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Taatjes, D.J.; Näär, A.M.; Andel, F.; Nogales, E.; Tjian, R. Structure, function, and activator-induced conformations of the CRSP coactivator. Science 2002, 295, 1058–1062. [Google Scholar] [CrossRef]
- Mo, X.; Kowenz-Leutz, E.; Xu, H.; Leutz, A. Ras Induces Mediator Complex Exchange on C/EBPβ. Mol. Cell 2004, 13, 241–250. [Google Scholar] [CrossRef]
- Pavri, R.; Lewis, B.; Kim, T.-K.; Dilworth, F.J.; Erdjument-Bromage, H.; Tempst, P.; de Murcia, G.; Evans, R.; Chambon, P.; Reinberg, D. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol. Cell 2005, 18, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, M.T.; Meyer, K.D.; Bernecky, C.; Taatjes, D.J. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009, 23, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebmeier, C.C.; Taatjes, D.J. Activator-Mediator binding regulates Mediator-cofactor interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 11283–11288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605. [Google Scholar] [CrossRef] [PubMed]
- Gaiano, N.; Fishell, G. The role of notch in promoting glial and neural stem cell fates. Annu. Rev. Neurosci. 2002, 25, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Laky, K.; Fowlkes, B.J. Notch signaling in CD4 and CD8 T cell development. Curr. Opin. Immunol. 2008, 20, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind Recruits CycC:CDK8 to Phosphorylate the Notch ICD and Coordinate Activation with Turnover. Mol. Cell 2004, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-Signaling from Cell Membrane to the Nucleus have been observed in both TGF-family receptors and the Smad proteins. The TGF-type II receptor is inacti-vated by mutation in most human gastrointestinal can. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Gao, S.; Alarcón, C.; Sapkota, G.; Rahman, S.; Chen, P.Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massagué, J. Ubiquitin Ligase Nedd4L Targets Activated Smad2/3 to Limit TGF-β Signaling. Mol. Cell 2009, 36, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, G.; Alarcón, C.; Spagnoli, F.M.; Brivanlou, A.H.; Massagué, J. Balancing BMP Signaling through Integrated Inputs into the Smad1 Linker. Mol. Cell 2007, 25, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.; Zaromytidou, A.; Xi, Q.; Gao, S.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-todorova, K.; Macias, J.; Sapkota, G.; et al. interactions in BMP and TGF β pathways. Cell 2010, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Sadzak, I.; Schiff, M.; Gattermeier, I.; Glinitzer, R.; Sauer, I.; Saalmüller, A.; Yang, E.; Schaljo, B.; Kovarik, P. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 8944–8949. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Darnell, J.E. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dölken, L.; Strobl, B.; Müller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef]
- Firestein, R.; Shima, K.; Nosho, K.; Irahara, N.; Baba, Y. CDK8 Expression in 470 Colorectal Cancers in Relation to ß-Catenin Activation, Other Molecular Alterations and Patient Survival. Int. J. Cancer 2011, 126, 2863–2873. [Google Scholar]
- Seo, J.-O.; Han, S.I.; Lim, S.-C. Role of CDK8 and beta-catenin in colorectal adenocarcinoma. Oncol. Rep. 2010, 24, 285–291. [Google Scholar]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-H.; Lee, S.-W.; Wang, J.; Lin, H.-K. Regulation of Skp2 expression and activity and its role in cancer progression. Sci. World J. 2010, 10, 1001–1015. [Google Scholar] [CrossRef]
- Xu, D.; Li, C.-F.; Zhang, X.; Gong, Z.; Chan, C.-H.; Lee, S.-W.; Jin, G.; Rezaeian, A.-H.; Han, F.; Wang, J.; et al. Skp2-MacroH2A1-CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat. Commun. 2015, 6, 6641. [Google Scholar] [CrossRef] [PubMed]
- Broude, E.V.; Győrffy, B.; Chumanevich, A.A.; Chen, M.; McDermott, M.S.J.; Shtutman, M.; Catroppo, J.F.; Roninson, I.B. Expression of CDK8 and CDK8-interacting Genes as Potential Biomarkers in Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.S.J.; Chumanevich, A.A.; Lim, C.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H.; et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017, 8, 12558–12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Wang, Z.; Zhang, W.; Qian, K.; Li, H.; Kong, D.; Li, Y.; Tang, Y. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/β-catenin signaling pathway. Cancer Lett. 2015, 356, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhong, C.; Frenkel, B.; Reddi, A.H.; Roy-Burman, P. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res. 2005, 65, 5769–5777. [Google Scholar] [CrossRef]
- Deng, H.; Makizumi, R.; Ravikumar, T.S.; Dong, H.; Yang, W.; Yang, W.L. Bone morphogenetic protein-4 is overexpressed in colonic adenocarcinomas and promotes migration and invasion of HCT116 cells. Exp. Cell Res. 2007, 313, 1033–1044. [Google Scholar] [CrossRef]
- Thériault, B.L.; Shepherd, T.G.; Mujoomdar, M.L.; Nachtigal, M.W. BMP4 induces EMT and Rho GTPase activation in human ovarian cancer cells. Carcinogenesis 2007, 28, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.J.; Kirkbride, K.C.; How, T.; Blobe, G.C. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2009, 30, 238–248. [Google Scholar] [CrossRef]
- Serrao, A.; Jenkins, L.M.; Chumanevich, A.A.; Horst, B.; Liang, J.; Gatza, M.L.; Lee, N.Y.; Roninson, I.B.; Broude, E.V.; Mythreye, K. Mediator kinase CDK8/CDK19 drives YAP1-dependent BMP4-induced EMT in cancer. Oncogene 2018, 37, 4792–4808. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Kong, L.; Xiao, Y.; Yuan, H.; Song, Y.; Wang, J.; Yu, H.; Mao, S.; Xu, W. CDK8 regulates the angiogenesis of pancreatic cancer cells in part via the CDK8-β-catenin-KLF2 signal axis. Exp. Cell Res. 2018, 369, 304–315. [Google Scholar] [CrossRef]
- Abdalla, E.K.; Adam, R.; Bilchik, A.J.; Jaeck, D.; Vauthey, J.-N.; Mahvi, D. Improving Resectability of Hepatic Colorectal Metastases: Expert Consensus Statement. Ann. Surg. Oncol. 2006, 13, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, M.; Hughes, D.; Chumanevich, A.A.; Altilia, S.; Kaza, V.; Lim, C.-U.; Kiaris, H.; Mythreye, K.; Pena, M.M.; et al. CDK8 selectively promotes the growth of colon cancer metastases in the liver by regulating gene expression of TIMP3 and matrix metalloproteinases. Cancer Res. 2018, 78, canres.1583.2018. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, Y.; Wang, H.; Xu, D.; Meng, X.; Shao, Y.; Lin, C.; Ye, Y.; Qian, H.; Wang, S. Tissue inhibitor of metalloproteinases-3 transfer suppresses malignant behaviors of colorectal cancer cells. Cancer Gene Ther. 2012, 19, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Brägelmann, J.; Klümper, N.; Offermann, A.; von Mässenhausen, A.; Böhm, D.; Deng, M.; Queisser, A.; Sanders, C.; Syring, I.; Merseburger, A.S.; et al. Pan-Cancer Analysis of the Mediator Complex Transcriptome Identifies CDK19 and CDK8 as Therapeutic Targets in Advanced Prostate Cancer. Clin. Cancer Res. 2017, 23, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Beer, P.A.; Godfrey, A.L.; Ortmann, C.A.; Li, J.; Costa-Pereira, A.P.; Ingle, C.E.; Dermitzakis, E.T.; Campbell, P.J.; Green, A.R. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell 2010, 18, 524–535. [Google Scholar] [CrossRef]
- Nitulescu, I.I.; Meyer, S.C.; Wen, Q.J.; Crispino, J.D.; Lemieux, M.E.; Levine, R.L.; Pelish, H.E.; Shair, M.D. Mediator Kinase Phosphorylation of STAT1 S727 Promotes Growth of Neoplasms With JAK-STAT Activation. EBioMedicine 2017, 26, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Rzymski, T.; Mikula, M.; Żyłkiewicz, E.; Dreas, A.; Wiklik, K.; Gołas, A.; Wójcik, K.; Masiejczyk, M.; Wróbel, A.; Dolata, I.; et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017, 8, 33779–33795. [Google Scholar] [CrossRef]
- Li, N.; Fassl, A.; Chick, J.; Inuzuka, H.; Li, X.; Mansour, M.R.; Liu, L.; Wang, H.; King, B.; Shaik, S.; et al. Cyclin C is a haploinsufficient tumour suppressor. Nat. Cell Biol. 2014, 16, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Gade, P.; Nallar, S.C.; Raha, A.; Roy, S.K.; Karra, S.; Reddy, J.K.; Reddy, S.P.; Kalvakolanu, D.V. The Med1 subunit of transcriptional mediator plays a central role in regulating CCAAT/enhancer-binding protein-β-driven transcription in response to interferon-γ. J. Biol. Chem. 2008, 283, 13077–13086. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, T.; Fukasawa, R.; Shinmyouzu, K.; Nakagawa, R.; Tobe, K.; Tanaka, A.; Ohkuma, Y. Mediator complex recruits epigenetic regulators via its two cyclin-dependent kinase subunits to repress transcription of immune response genes. J. Biol. Chem. 2013, 288, 20955–20965. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Hagihara, T.; Horiuchi, Y.; Okui, A.; Wani, S.; Yoshida, T.; Inoue, T.; Tanaka, A.; Ito, T.; Hirose, Y.; et al. Mediator cyclin-dependent kinases upregulate transcription of inflammatory genes in cooperation with NF-κB and C/EBPβ on stimulation of Toll-like receptor 9. Genes to Cells 2017, 22, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Gotthardt, D.; Hoermann, G.; Csiszar, A.; Wirth, S.; Berger, A.; Straka, E.; Rigler, D.; Wallner, B.; Jamieson, A.M.; et al. CDK8-Mediated STAT1-S727 Phosphorylation Restrains NK Cell Cytotoxicity and Tumor Surveillance. Cell Rep. 2013, 4, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckelhart, E.; Warsch, W.; Zebedin, E.; Simma, O.; Stoiber, D.; Kolbe, T.; Rülicke, T.; Mueller, M.; Casanova, E.; Sexl, V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011, 117, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witalisz-Siepracka, A.; Gotthardt, D.; Prchal-Murphy, M.; Didara, Z.; Menzl, I.; Prinz, D.; Edlinger, L.; Putz, E.M.; Sexl, V. NK Cell–Specific CDK8 Deletion Enhances Antitumor Responses. Cancer Immunol. Res. 2018, 6, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E.I. Killing-Off of Tumor Cells in Vitro. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C. V Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Andrysik, Z.; Pandey, A.; Hoh, M.; Bonner, E.A.; Hill, A.A.; Sullivan, K.D.; Espinosa, J.M. CDK8 Kinase Activity Promotes Glycolysis. Cell Rep. 2017, 21, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Feng, D.; Wang, Q.; Abdulla, A.; Xie, X.J.; Zhou, J.; Sun, Y.; Yang, E.S.; Liu, L.P.; Vaitheesvaran, B.; et al. Regulation of lipogenesis by cyclin-dependent kinase 8 - Mediated control of SREBP-1. J. Clin. Investig. 2012, 122, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Youn, D.Y.; Zhao, X.; Gao, Y.; Quinn, W.J.; Xiaoli, A.M.; Sun, Y.; Birnbaum, M.J.; Pessin, J.E.; Yang, F. mTORC1 down-regulates cyclin-dependent kinase 8 (CDK8) and cyclin C (CycC). PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menzl, I.; Witalisz-Siepracka, A.; Sexl, V. CDK8-Novel Therapeutic Opportunities. Pharmaceuticals 2019, 12, 92. https://doi.org/10.3390/ph12020092
Menzl I, Witalisz-Siepracka A, Sexl V. CDK8-Novel Therapeutic Opportunities. Pharmaceuticals. 2019; 12(2):92. https://doi.org/10.3390/ph12020092
Chicago/Turabian StyleMenzl, Ingeborg, Agnieszka Witalisz-Siepracka, and Veronika Sexl. 2019. "CDK8-Novel Therapeutic Opportunities" Pharmaceuticals 12, no. 2: 92. https://doi.org/10.3390/ph12020092
APA StyleMenzl, I., Witalisz-Siepracka, A., & Sexl, V. (2019). CDK8-Novel Therapeutic Opportunities. Pharmaceuticals, 12(2), 92. https://doi.org/10.3390/ph12020092