HFE Related Hemochromatosis: Uncovering the Inextricable Link between Iron Homeostasis and the Immunological System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Learning: A Brief Historical Sequence Where the Improbable Led to Discovery
2. Results
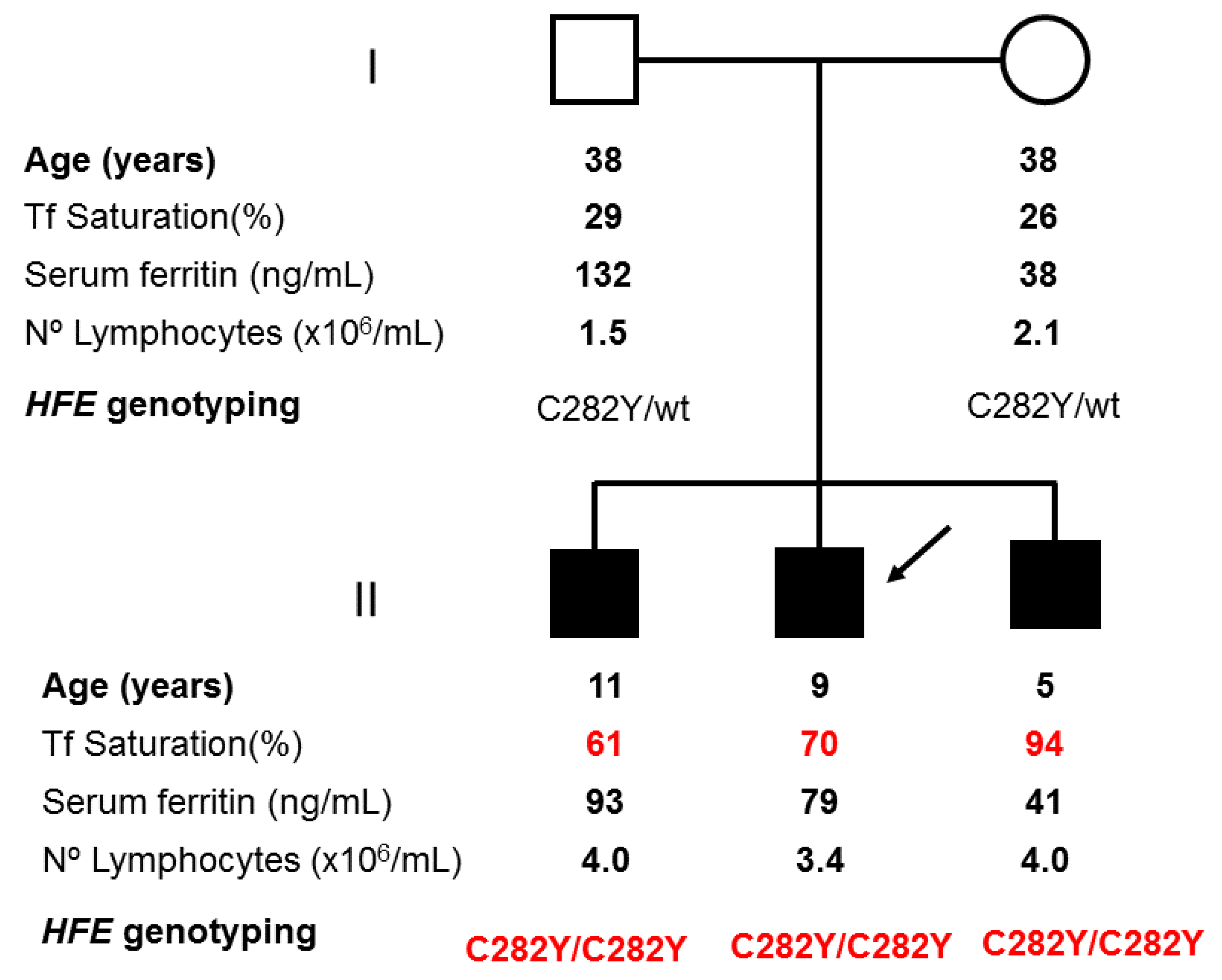
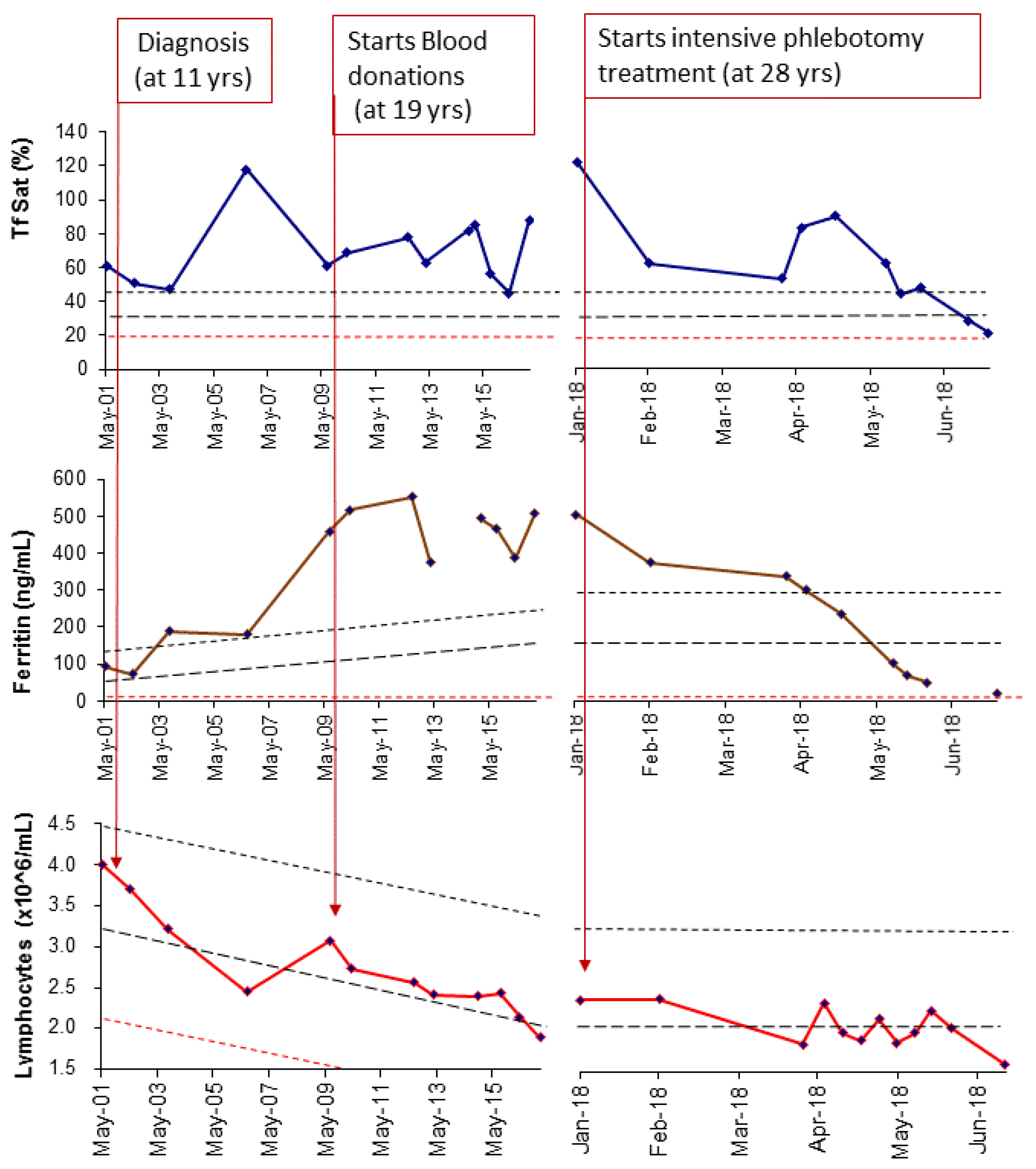
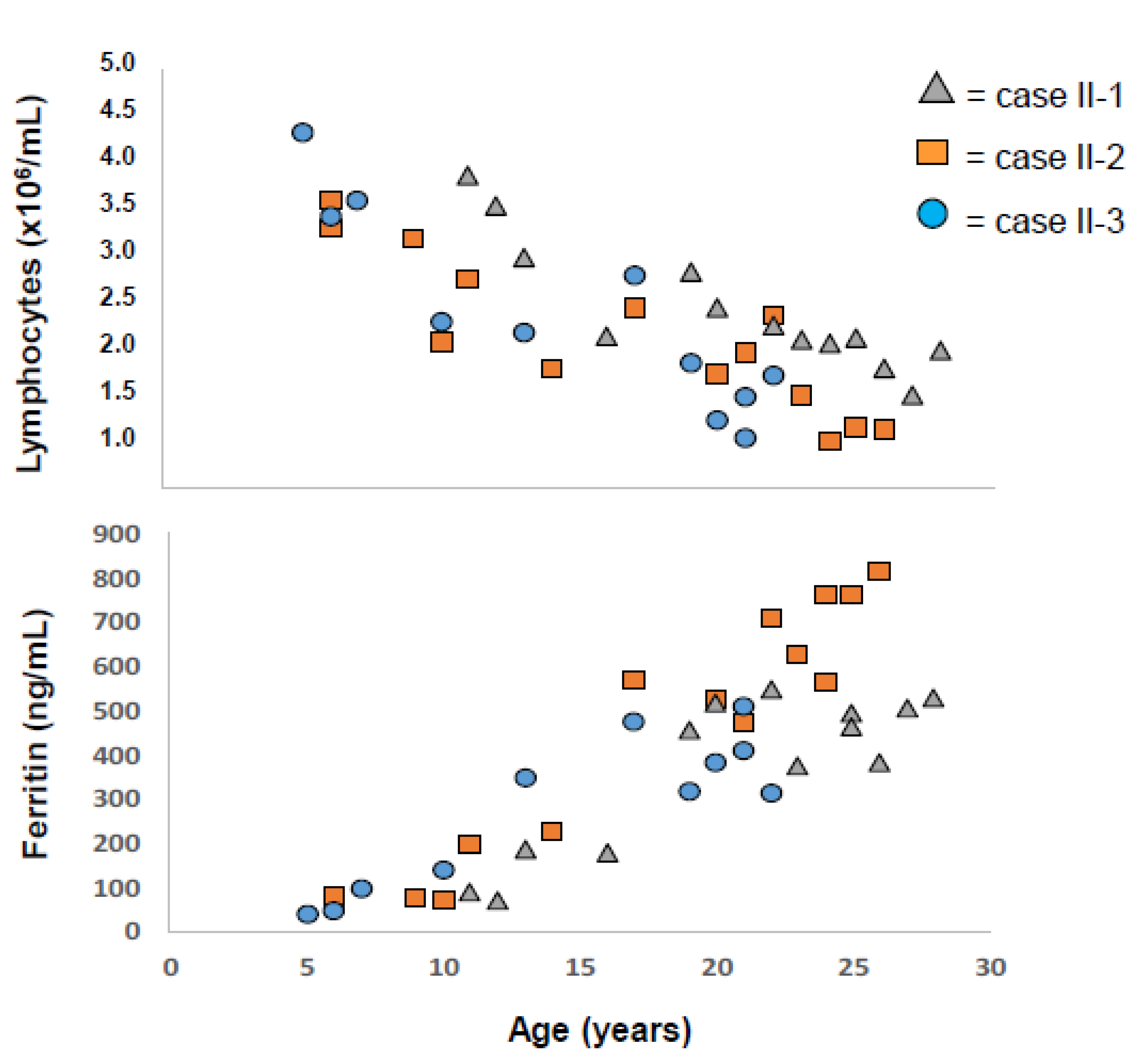
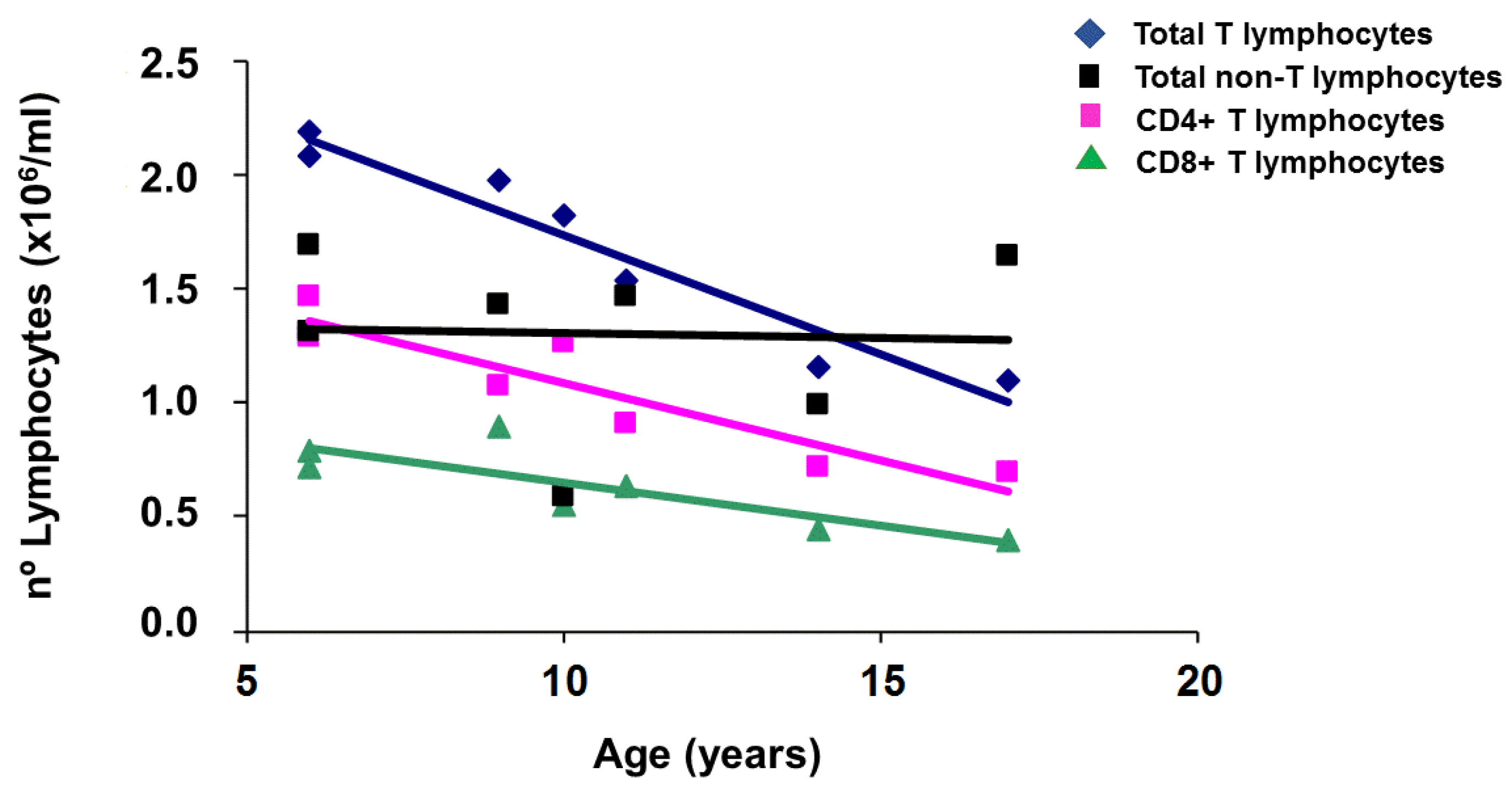
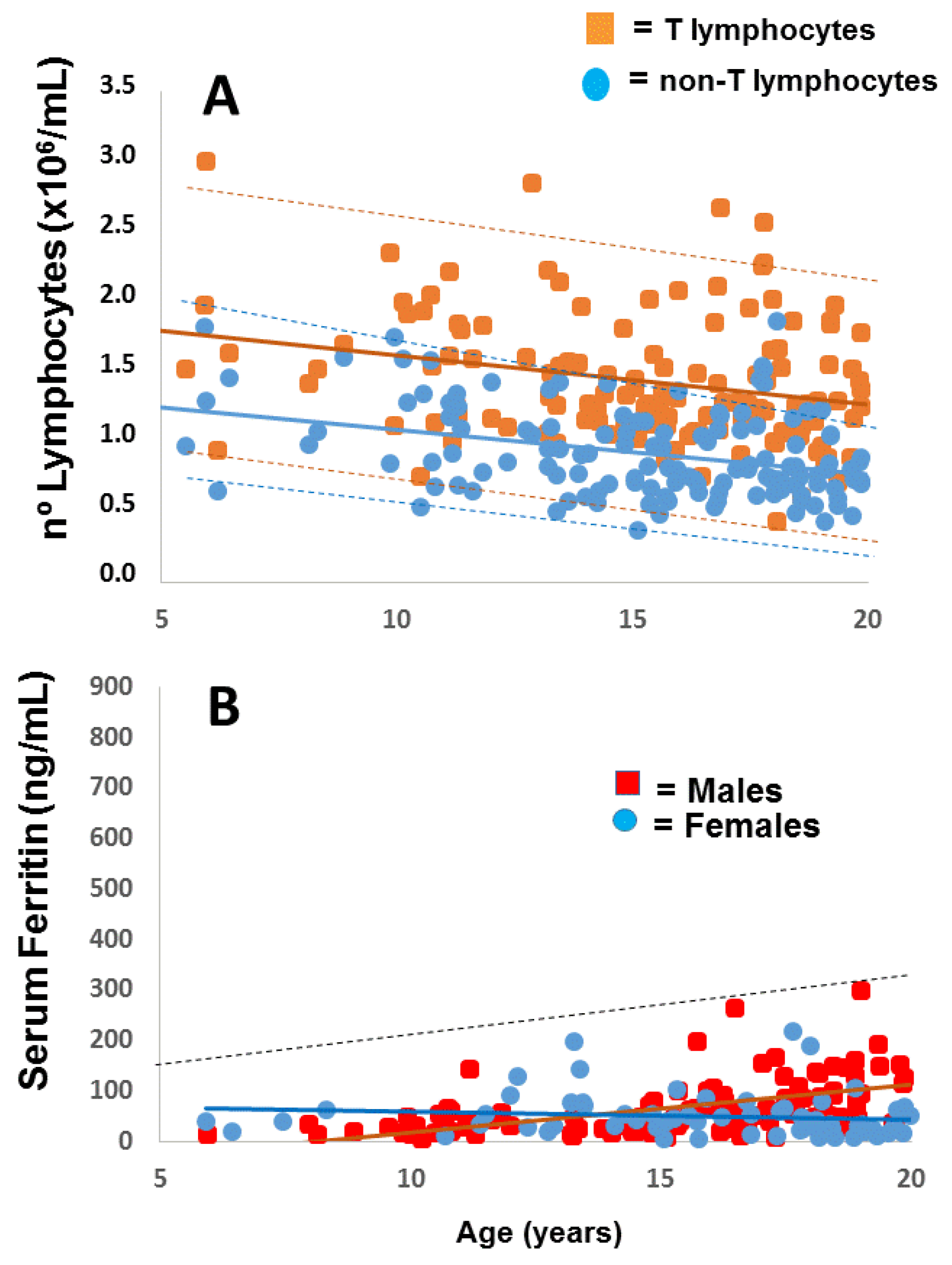
2.1. Applying: A Case and Family Report
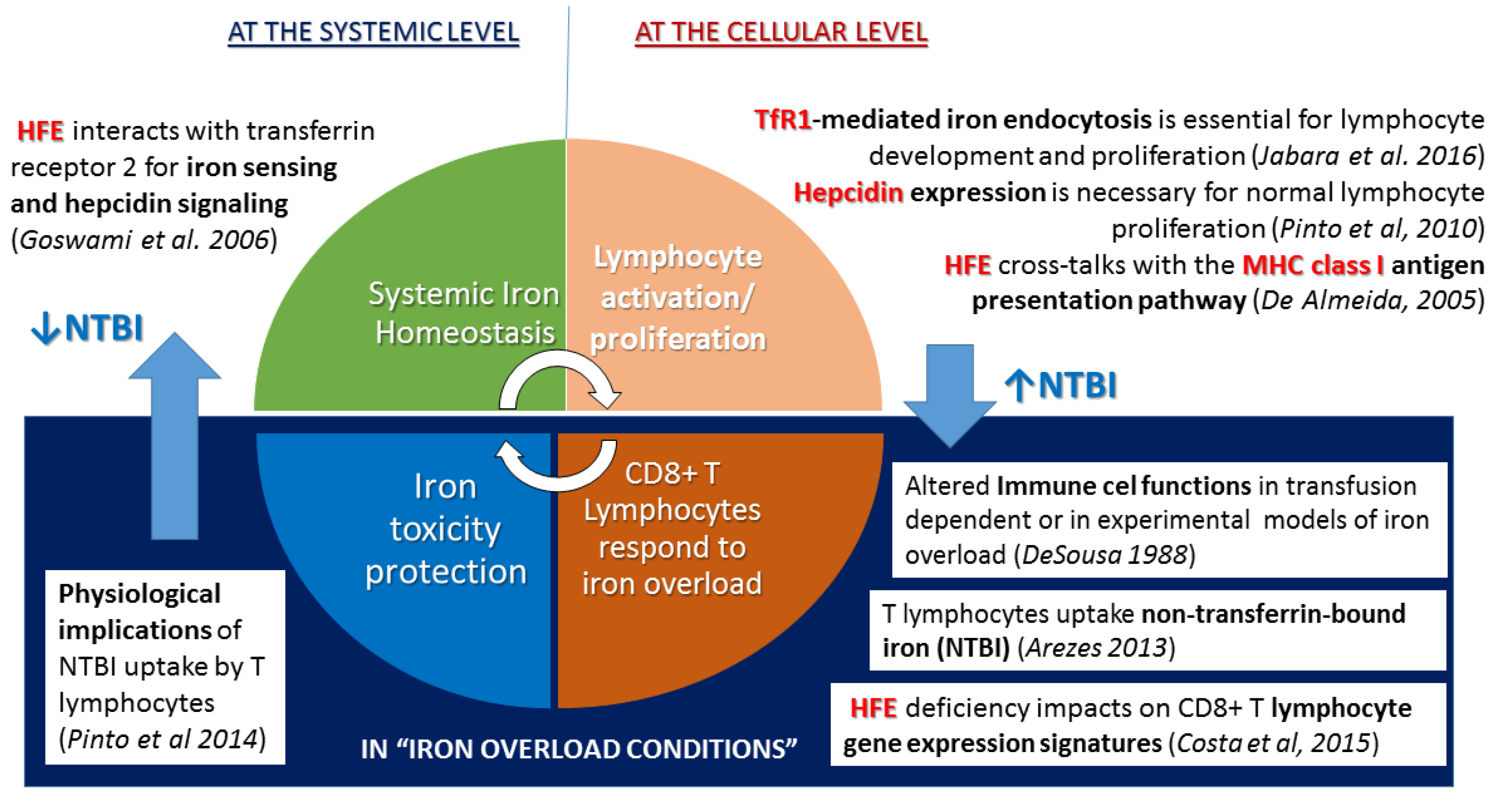
2.2. Questioning: A Dual Function for HFE?
3. Discussion
3.1. Implications for Disease Prevention in HFE-Related Hemochromatosis
3.2. Implications for Immunological Theory
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adams, P.C.; Barton, J.C. Haemochromatosis. Lancet 2007, 370, 1855–1860. [Google Scholar] [CrossRef]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; Graaff, B.; McLaren, C.E.; Loreal, O. Haemochromatosis. Nat. Revs. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochimica Et Biophysica Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Almeida, S.F.; Carvalho, I.F.; Cardoso, C.S.; Cordeiro, J.V.; Azevedo, J.E.; Neefjes, J.; de Sousa, M. HFE cross-talks with the MHC class I antigen presentation pathway. Blood 2005, 106, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Rohrlich, P.S.; Kanellopoulos, J.; Lemonnier, F.A. HFE, a MHC class Ib molecule that regulates iron metabolism. Med. Sci. M/S 2006, 22, 24–26. [Google Scholar] [PubMed]
- Reuben, A.; Chung, J.W.; Lapointe, R.; Santos, M.M. The hemochromatosis protein HFE 20 years later: An emerging role in antigen presentation and in the immune system. Immun. Inflamm. Dis. 2017, 5, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Arosa, F.A.; Oliveira, L.; Porto, G.; da Silva, B.M.; Kruijer, W.; Veltman, J.; de Sousa, M. Anomalies of the CD8+ T cell pool in haemochromatosis: HLA-A3-linked expansions of CD8+CD28− T cells. Clin. Exp. Immunol. 1997, 107, 548–554. [Google Scholar] [CrossRef]
- Porto, G.; Reimao, R.; Goncalves, C.; Vicente, C.; Justica, B.; de Sousa, M. Haemochromatosis as a window into the study of the immunological system: A novel correlation between CD8+ lymphocytes and iron overload. Eur. J. Haematol. 1994, 52, 283–290. [Google Scholar] [CrossRef]
- Pinto, J.P.; Arezes, J.; Dias, V.; Oliveira, S.; Vieira, I.; Costa, M.; Vos, M.; Carlsson, A.; Rikers, Y.; Rangel, M.; et al. Physiological implications of NTBI uptake by T lymphocytes. Frontiers. Pharmacol. 2014, 5, 24. [Google Scholar]
- Simon, M.; Pawlotsky, Y.; Bourel, M.; Fauchet, R.; Genetet, B. Letter: Idiopathic hemochromatosis associated with HL-A 3 tissular antigen. Nouv. Presse Med. 1975, 4, 1432. [Google Scholar] [PubMed]
- Simon, M.; Bourel, M.; Fauchet, R.; Genetet, B. HLA and "non-immunological" disease: Idiopathic haemochromatosis. Lancet 1976, 308, 973–974. [Google Scholar] [CrossRef]
- de Sousa, M. Lymphoid cell positioning: A new proposal for the mechanism of control of lymphoid cell migration. Sympo. Soc. Exp. Biol. 1978, 32, 393–410. [Google Scholar]
- Reimao, R.; Porto, G.; de Sousa, M. Stability of CD4/CD8 ratios in man: New correlation between CD4/CD8 profiles and iron overload in idiopathic haemochromatosis patients. C. R. A. Sci. 1991, 313, 481–487. [Google Scholar]
- Porto, G.; Vicente, C.; Teixeira, M.A.; Martins, O.; Cabeda, J.M.; Lacerda, R.; Goncalves, C.; Fraga, J.; Macedo, G.; Silva, B.M.; et al. Relative impact of HLA phenotype and CD4-CD8 ratios on the clinical expression of hemochromatosis. Hepatology 1997, 25, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Deugnier, Y.; Morcet, J.; Laine, F.; Hamdi-Roze, H.; Bollard, A.S.; Guyader, D.; Moirand, R.; Bardou-Jacquet, E. Reduced phenotypic expression in genetic hemochromatosis with time: Role of exposure to non-genetic modifiers. J. Hepatol. 2019, 70, 118–125. [Google Scholar] [CrossRef]
- Cruz, E.; Vieira, J.; Almeida, S.; Lacerda, R.; Gartner, A.; Cardoso, C.S.; Alves, H.; Porto, G. A study of 82 extended HLA haplotypes in HFE-C282Y homozygous hemochromatosis subjects: Relationship to the genetic control of CD8+ T-lymphocyte numbers and severity of iron overload. BMC MED. GENET. 2006, 7, 16. [Google Scholar] [CrossRef]
- Costa, M.; Cruz, E.; Barton, J.C.; Thorstensen, K.; Morais, S.; da Silva, B.M.; Pinto, J.P.; Vieira, C.P.; Vieira, J.; Acton, R.T.; et al. Effects of highly conserved major histocompatibility complex (MHC) extended haplotypes on iron and low CD8+ T lymphocyte phenotypes in HFE C282Y homozygous hemochromatosis patients from three geographically distant areas. PloS ONE 2013, 8, e79990. [Google Scholar] [CrossRef]
- Cruz, E.; Melo, G.; Lacerda, R.; Almeida, S.; Porto, G. The CD8+ T-lymphocyte profile as a modifier of iron overload in HFE hemochromatosis: An update of clinical and immunological data from 70 C282Y homozygous subjects. Blood Cells Mol. Dis. 2006, 37, 33–39. [Google Scholar] [CrossRef]
- Macedo, M.F.; Porto, G.; Costa, M.; Vieira, C.P.; Rocha, B.; Cruz, E. Low numbers of CD8+ T lymphocytes in hereditary haemochromatosis are explained by a decrease of the most mature CD8+ effector memory T cells. Clin. Exp. Immunol. 2010, 159, 363–371. [Google Scholar] [CrossRef]
- de Sousa, M.; Dynesius-Trentham, R.; Mota-Garcia, F.; da Silva, M.T.; Trentham, D.E. Activation of rat synovium by iron. Arthritis Rheum. 1988, 31, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, F.C.; Dynesius-Trentham, R.; de Sousa, M.; Trentham, D.E. Collagen arthritis in the rat is initiated by CD4+ T cells and can be amplified by iron. Cell. Immunol. 1989, 121, 1–12. [Google Scholar] [CrossRef]
- de Sousa, M. Immune cell functions in iron overload. Clin. Exp. Immunol. 1989, 75, 1–6. [Google Scholar] [PubMed]
- de Sousa, M.; Reimao, R.; Porto, G.; Grady, R.W.; Hilgartner, M.W.; Giardina, P. Iron and lymphocytes: Reciprocal regulatory interactions. In Biotechnology of Plasma Proteins; Karger Publishers: Berlin, Germany, 1991; Volume 58, pp. 171–177. [Google Scholar]
- de Sousa, M.; Reimao, R.; Lacerda, R.; Hugo, P.; Kaufmann, S.H.; Porto, G. Iron overload in β 2-microglobulin-deficient mice. Immunol. Lett. 1994, 39, 105–111. [Google Scholar] [CrossRef]
- Santos, M.; Schilham, M.W.; Rademakers, L.H.; Marx, J.J.; de Sousa, M.; Clevers, H. Defective iron homeostasis in β 2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J. Exp. Med. 1996, 184, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rodrigues, P.; Macedo, M.G.; Minana, B.; Brennan, K.; Cardoso, E.M.; Hentze, M.W.; de Sousa, M. Molecular analysis of iron overload in β2-microglobulin-deficient mice. Blood Cells Mol. Dis. 2004, 33, 125–131. [Google Scholar] [CrossRef]
- Santos, M.M.; de Sousa, M.; Rademakers, L.H.; Clevers, H.; Marx, J.J.; Schilham, M.W. Iron overload and heart fibrosis in mice deficient for both β2-microglobulin and Rag1. Am. J. Pathol. 2000, 157, 1883–1892. [Google Scholar] [CrossRef]
- Levy, J.E.; Montross, L.K.; Andrews, N.C. Genes that modify the hemochromatosis phenotype in mice. J. Clin. Invest. 2000, 105, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, E.M.; Macedo, M.G.; Rohrlich, P.; Ribeiro, E.; Silva, M.T.; Lemonnier, F.A.; de Sousa, M. Increased hepatic iron in mice lacking classical MHC class I molecules. Blood 2002, 100, 4239–4241. [Google Scholar] [CrossRef]
- Costa, M.; Cruz, E.; Oliveira, S.; Benes, V.; Ivacevic, T.; Silva, M.J.; Vieira, I.; Dias, F.; Fonseca, S.; Gonçalves, M.; et al. Lymphocyte gene expression signatures from patients and mouse models of hereditary hemochromatosis reveal a function of HFE as a negative regulator of CD8+ T-lymphocyte activation and differentiation in vivo. PLoS ONE 2015, 10, e0124246. [Google Scholar] [CrossRef]
- Arezes, J.; Costa, M.; Vieira, I.; Dias, V.; Kong, X.L.; Fernandes, R.; Vos, M.; Carlsson, A.; Rikers, Y.; Porto, G.; et al. Non-transferrin-bound iron (NTBI) uptake by T lymphocytes: Evidence for the selective acquisition of oligomeric ferric citrate species. PLoS ONE 2013, 8, e79870. [Google Scholar] [CrossRef]
- Jabara, H.H.; Boyden, S.E.; Chou, J.; Ramesh, N.; Massaad, M.J.; Benson, H.; Bainter, W.; Fraulino, D.; Rahimov, F.; Sieff, C.; et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 2016, 48, 74–78. [Google Scholar] [CrossRef]
- Vanoaica, L.; Richman, L.; Jaworski, M.; Darshan, D.; Luther, S.A.; Kuhn, L.C. Conditional deletion of ferritin h in mice reduces B and T lymphocyte populations. PLoS ONE 2014, 9, e89270. [Google Scholar] [CrossRef]
- Pinto, J.P.; Dias, V.; Zoller, H.; Porto, G.; Carmo, H.; Carvalho, F.; de Sousa, M. Hepcidin messenger RNA expression in human lymphocytes. Immunology 2010, 130, 217–230. [Google Scholar] [CrossRef]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef]
- Moura, E.; Noordermeer, M.A.; Verhoeven, N.; Verheul, A.F.; Marx, J.J. Iron release from human monocytes after erythrophagocytosis in vitro: An investigation in normal subjects and hereditary hemochromatosis patients. Blood 1998, 92, 2511–2519. [Google Scholar]
- Nnah, I.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 19. [Google Scholar]
- Available online: https://jamanetwork.com/journals/jama/fullarticle/1760318 (accessed on 27 November 2013).
- Svejgaard, A.; Ryder, L.P. Interaction of HLA molecules with non-immunological ligands as an explanation of HLA and disease associations. Lancet 1976, 2, 547–549. [Google Scholar] [CrossRef]
- Roy, C.N.; Penny, D.M.; Feder, J.N.; Enns, C.A. The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J. Biol. Chems. 1999, 274, 9022–9028. [Google Scholar] [CrossRef]
- Goswami, T.; Andrews, N.C. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chems. 2006, 281, 28494–28498. [Google Scholar] [CrossRef]
- Boucherma, R.; Kridane-Miledi, H.; Vives, F.L.; Vauchy, C.; Borg, C.; Kleinclauss, F.; Fiette, L.; Tiberghien, P.; Lemonnier, F.A.; Rohrlich, P.S.; et al. Loss of central and peripheral CD8+ T-cell tolerance to HFE in mouse models of human familial hemochromatosis. Eur J. Immunol. 2012, 42, 851–862. [Google Scholar] [CrossRef]
- de Almeida, S.F.; de Sousa, M. The unfolded protein response in hereditary haemochromatosis. J. Cell. Mol. Med. 2008, 12, 421–434. [Google Scholar] [CrossRef] [Green Version]
- Barton, J.C.; Acton, R.T.; Leiendecker-Foster, C.; Lovato, L.; Adams, P.C.; McLaren, G.D.; Eckfeldt, J.H.; McLaren, C.E.; Reboussin, D.M.; Gordeuk, V.R.; et al. HFE C282Y homozygotes aged 25–29 years at HEIRS Study initial screening. Genet. Test. 2007, 11, 269–275. [Google Scholar] [CrossRef]
- Andersen, R.V.; Tybjaerg-Hansen, A.; Appleyard, M.; Birgens, H.; Nordestgaard, B.G. Hemochromatosis mutations in the general population: Iron overload progression rate. Blood 2004, 103, 2914–2919. [Google Scholar] [CrossRef]
- Allen, K.J.; Bertalli, N.A.; Osborne, N.J.; Constantine, C.C.; Delatycki, M.B.; Nisselle, A.E.; Nicoll, A.J.; Gertig, D.M.; McLaren, C.E.; Giles, G.G.; et al. HFE Cys282Tyr homozygotes with serum ferritin concentrations below 1000 microg/L are at low risk of hemochromatosis. Hepatology 2010, 52, 925–933. [Google Scholar] [CrossRef]
- Powell, L.W.; Dixon, J.L.; Ramm, G.A.; Purdie, D.M.; Lincoln, D.J.; Anderson, G.J.; Subramaniam, V.N.; Hewett, D.G.; Searle, J.W.; Fletcher, L.M.; et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch. Intern. Med. 2006, 166, 294–301. [Google Scholar] [CrossRef]
- Soares, M.P.; Weiss, G. The Iron age of host-microbe interactions. EMBO Rep. 2015, 16, 1482–1500. [Google Scholar] [CrossRef]
- Rodrigues, P.; Lopes, C.; Mascarenhas, C.; Arosio, P.; Porto, G.; de Sousa, M. Comparative study between Hfe−/−and β2m−/− mice: Progression with age of iron status and liver pathology. Int. J. Exp. Pathol. 2006, 87, 317–324. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porto, G.; Cruz, E.; Teles, M.J.; de Sousa, M. HFE Related Hemochromatosis: Uncovering the Inextricable Link between Iron Homeostasis and the Immunological System. Pharmaceuticals 2019, 12, 122. https://doi.org/10.3390/ph12030122
Porto G, Cruz E, Teles MJ, de Sousa M. HFE Related Hemochromatosis: Uncovering the Inextricable Link between Iron Homeostasis and the Immunological System. Pharmaceuticals. 2019; 12(3):122. https://doi.org/10.3390/ph12030122
Chicago/Turabian StylePorto, Graça, Eugénia Cruz, Maria José Teles, and Maria de Sousa. 2019. "HFE Related Hemochromatosis: Uncovering the Inextricable Link between Iron Homeostasis and the Immunological System" Pharmaceuticals 12, no. 3: 122. https://doi.org/10.3390/ph12030122
APA StylePorto, G., Cruz, E., Teles, M. J., & de Sousa, M. (2019). HFE Related Hemochromatosis: Uncovering the Inextricable Link between Iron Homeostasis and the Immunological System. Pharmaceuticals, 12(3), 122. https://doi.org/10.3390/ph12030122